Filtering frames using a distance restraint
You want to filter certain frames from a trajectory for further post-processing.
In a ligand-receptor system, the ligand could be exploring different regions of the receptor over time. In order to select frames that contain a specific ligand position in respect to the receptor, we need to extract those frames using a certain type of cut-off. In this example, a 12-mer DNA is in contact with two ligands.
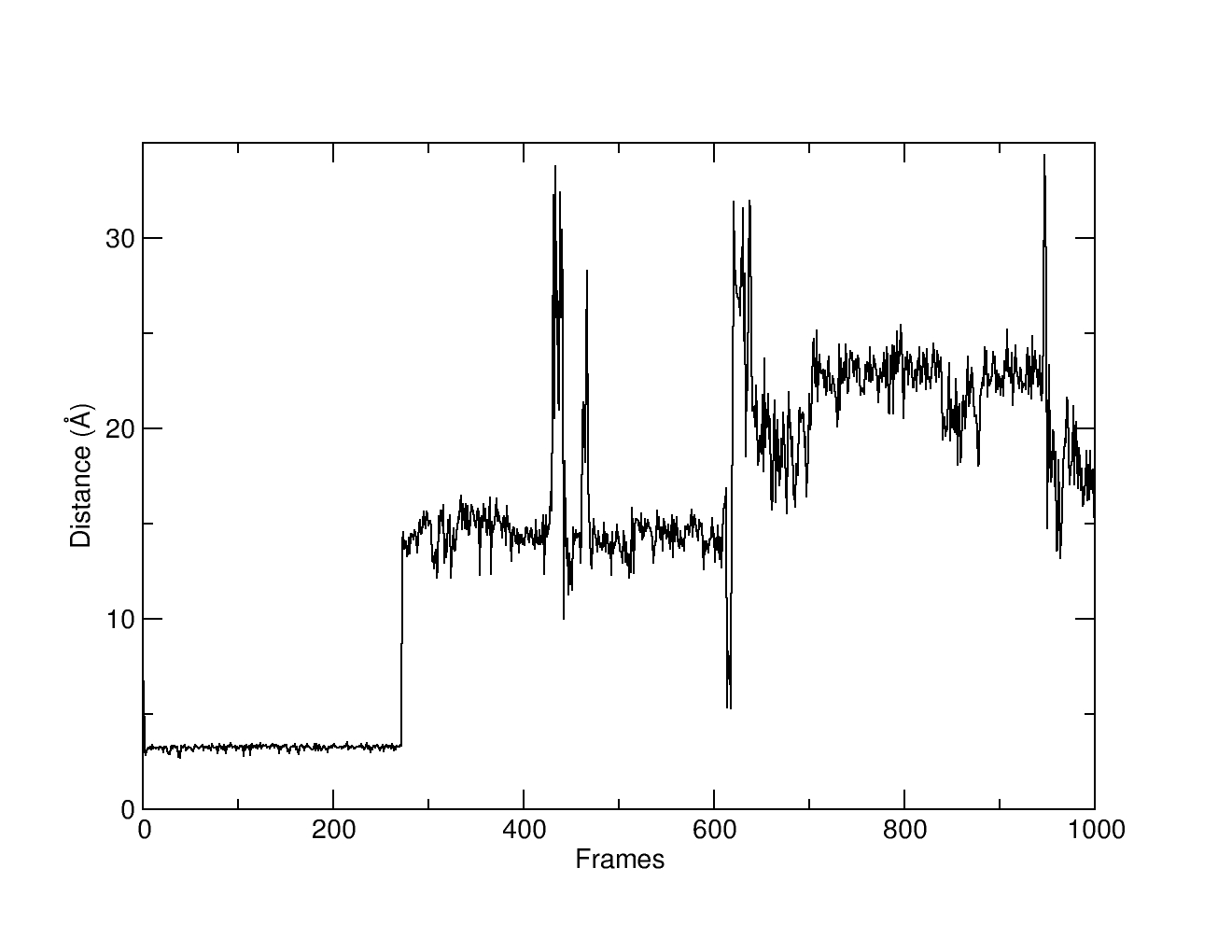
We can see from the movie (1000 frames trajectory) that the ligands are forming interactions between the phosphate oxygen atoms of the backbone and the copper center of the ligand. To further analyze those conformers, we want to extract the frames that have the ligand-DNA interaction. We can use the filter command to filter the distance between two atoms.
CPPTRAJ input:
parm vacuo.topo trajin vacuo.nc 1 last 1 rms fit :2-10,14-23 distance d1 :25@Cu1 :21@P out distance1.dat filter d1 min 2.0 max 4.0 out filter.dat outtraj distance1-filtered.nc netcdf distance d2 :25@Cu1 :21@P out distance2.dat go
The script reads the vacuo.topo topology and the vacuo.nc trajectory data from frame 1 to the last frame with a 1 frame off-set. The rms fit command will remove global rotational/translational motions in the residues 2-10 and 14-23. The distance command will have a dataset name of d1 and will read the distance between the atom name Cu1 in residue 25 (the ligand) and the phosphate atom (P) in residue 21. It will generate a file called distance1.dat with this data.
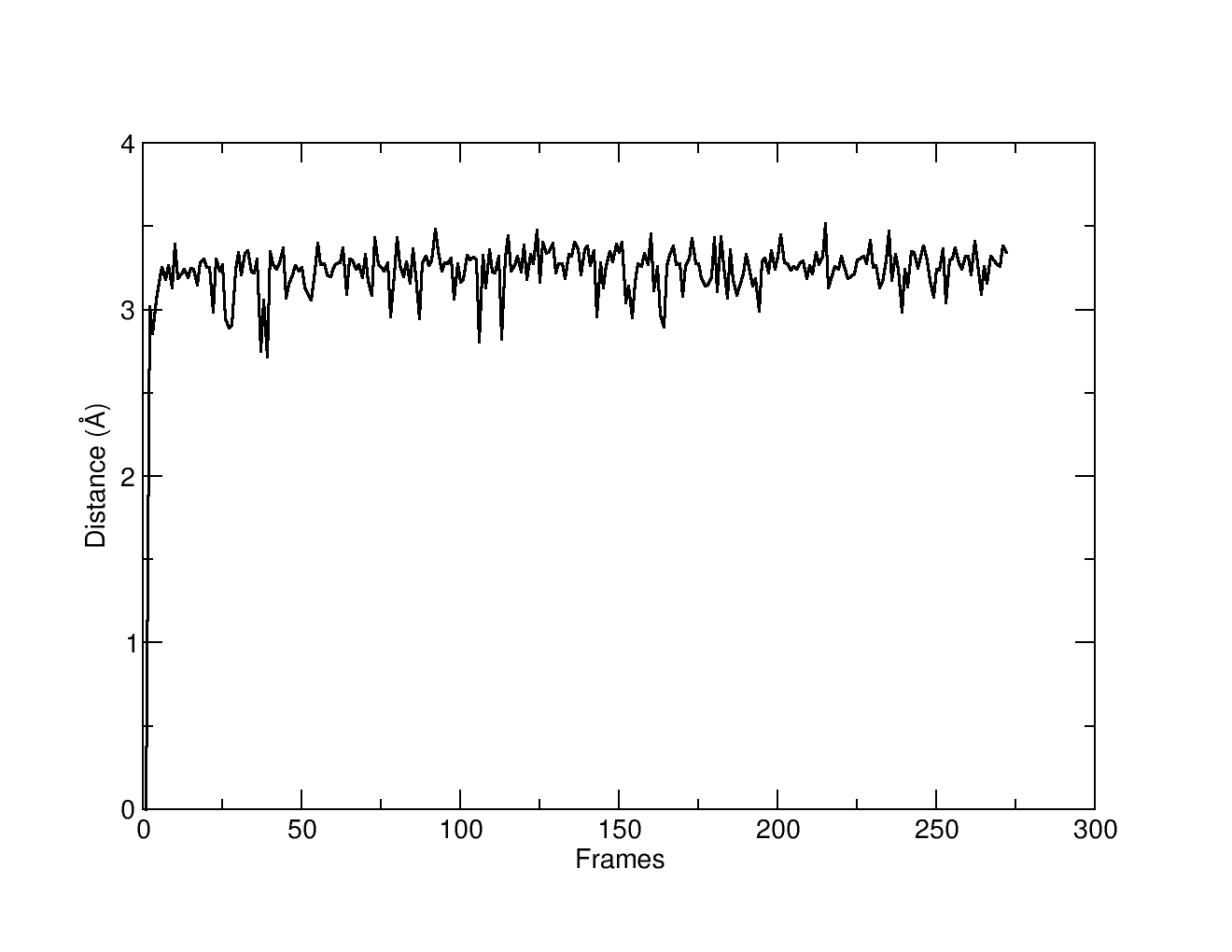
If we plot the distance1.dat, we can see that early in the simulation, the ligand spends ~270 frames withiin a distance of ~2.5-3.7 angstroms of the oxygen atoms in the phosphate of residue 25. We want to extract those frame for further processing (like measuring binding energy or analyzing the conformation of the ligand)
Using the filter command, we can read the output from the distance command (calling the dataset d1) and filter the frames that are within a minimim value of 2.5 and a maximum value of 4.0 angstroms. The filter command will extract the frames that fall within the desired filter. The results are stored in the filter.dat file that marks with a 1 if the frame fall within the filter’s range and with a 0 if it does not.
For the first 10 frames of filter.dat:
#Frame Filter_00003 1 0 2 1 3 1 4 1 5 1 6 1 7 1 8 1 9 1 10 1 11 1
At this stage, the trajectory file has been sieved and we can output this new trajectory using the outtraj command. The generated trajectory: distance1-filtered.nc will only contain 271 frames, that correspond to the frames where the ligand is within 2.0-4.0 angstroms from the phosphate atom. We can check that running a second distance command with dateset name ‘d2’ that will measure the same atoms as the ‘d1’ command. If we plot distance2.dat we can see that only the frames that pass through the filter command are now present: