
Simulation of a 10-mer DNA with a deoxy-5-methylcytosine residue
Incorporating a modified base (deoxy-5-methylcytosine) into DNA for MD simulation in AMBER.
For this example, we are going to incorportate a modified nucleotide (5-Methylcytosine) into the DNA duplex with the sequence d(CGCGCGCGCG)2 at posiiton 3. The result will be a double strand DNA with the sequence d(CG–DMC–GCGCGCG)-(GCGCGCGCGC), where DMC is the modified cytosine residue.
To start, we create our DNA duplex. There are multiple methods to generate a starting structure of nucleic acids or we can use an experimental structure from the Protein Data Bank. For our example we are going to use BIOVIA Discovery Studio Visualizer to create our starting structure.
Go to File->New-> Molecule Window.
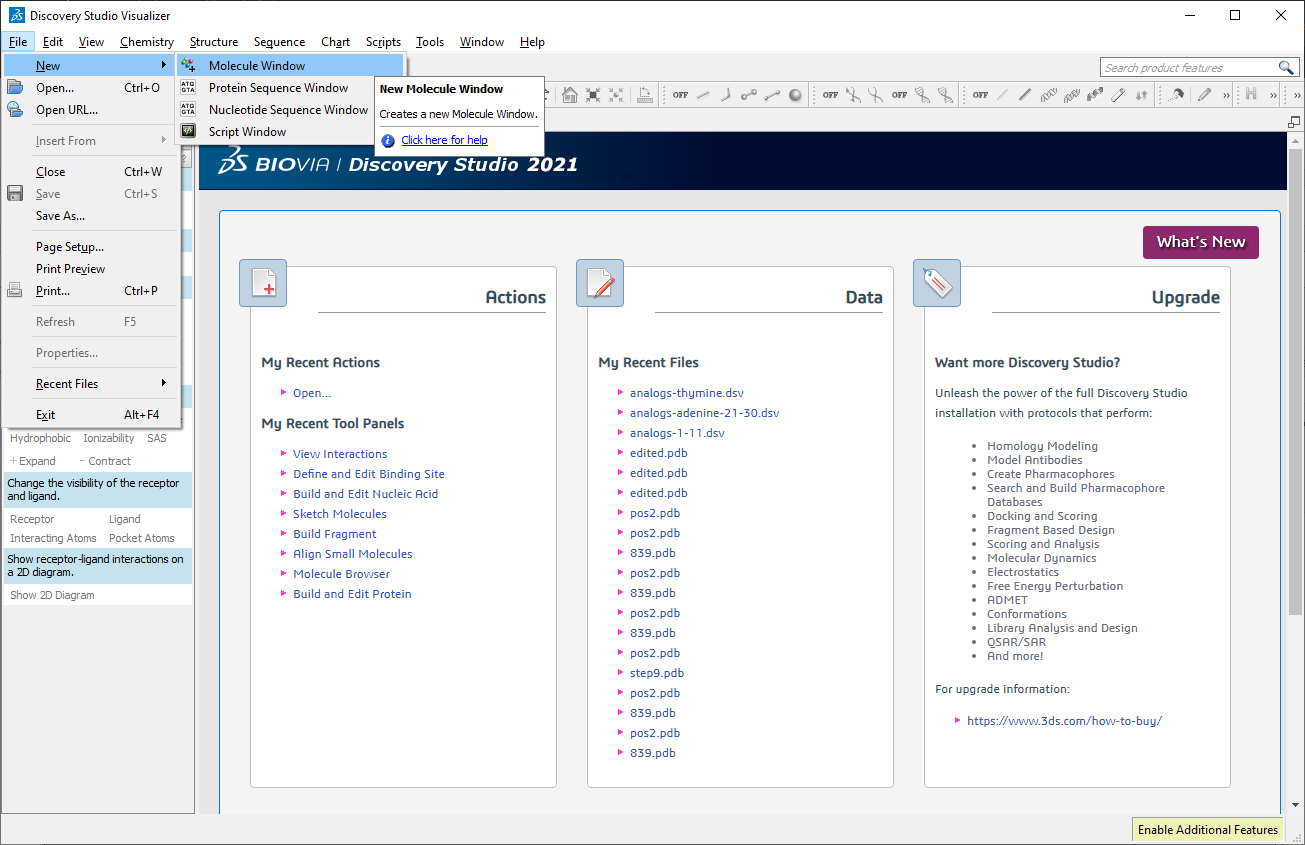
Build the DNA duplex. Go to Tools->Macromolecules->Build and Edit Nucleic Acid
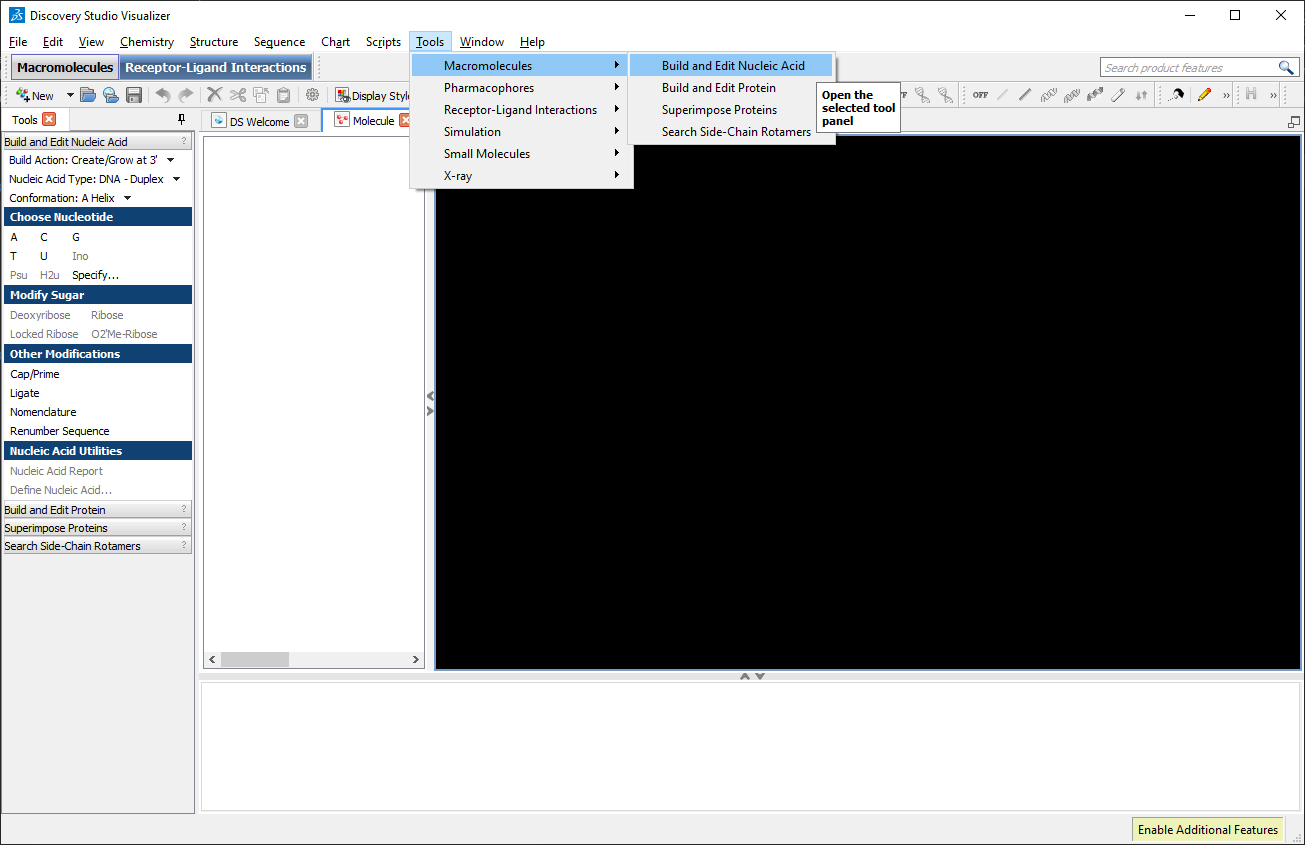
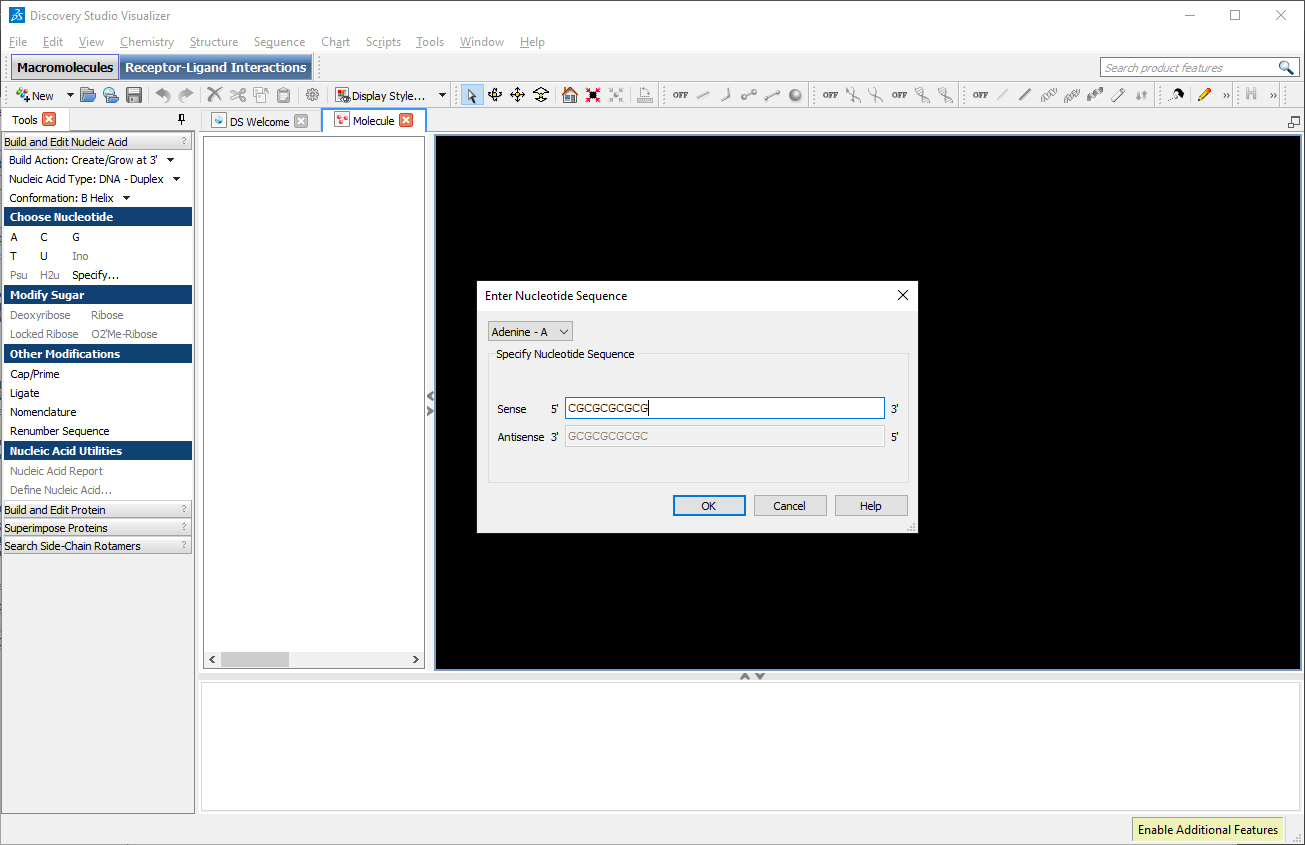
In the left toolbar, select “Build Action: Create/Grow at 3′”, in “Nucleic Acid Type: DNA – Duplex”, in “Build and Edit Nucleic Acid: Conformation: B Helix” and in “Choose Nucleotide” click on “Specify…”. A window will appear where we can insert the desired sequence. In the field Sense, type the desired sequence: CGCGCGCGCG.
This will generate our B-conformation DNA-duplex structure with the desired sequence (use right-click to rotate, scroll wheel to zoom in/out).
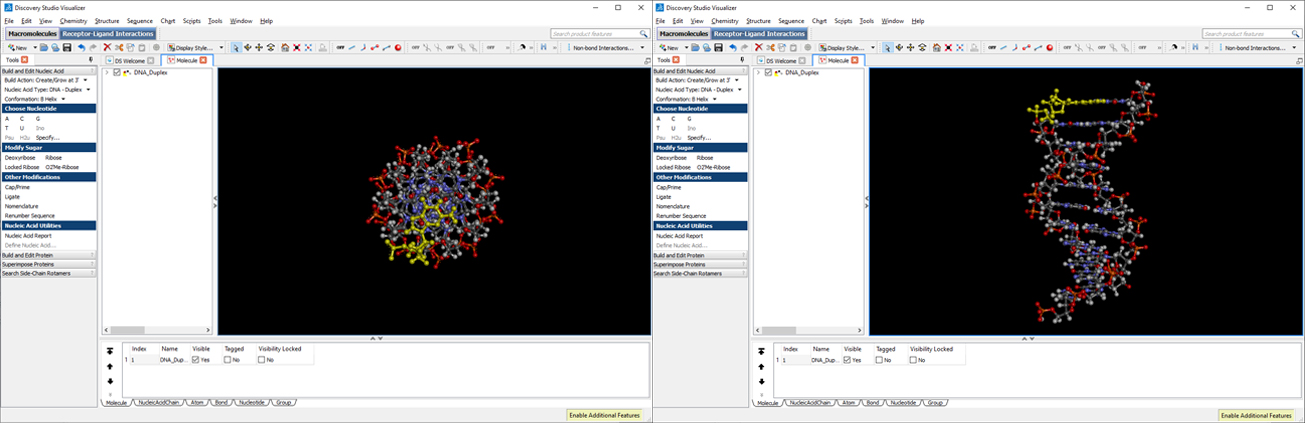
Click File->Save As… and save the file as dna.pdb (select PDB in the “Save as type:” dropdown menu). Download the dna.pdb file.
Now we need to incorporate our modified cytosine residue into our new DNA-duplex. To perform an AMBER Molecular Dynamics simulations, we require to have the proper force-field parameters (bonded, non-bonded and charges). For standard DNA and RNA residues, the parameters are already available in the force-fields included in Ambertools. For non-canonical DNA or RNA residues, we need to either generate the parameters or use parameters available in the literature. In this case, the deoxy-5-methylcytosine parameters are available from the AMBER parameter database curated by the Bryce Group.
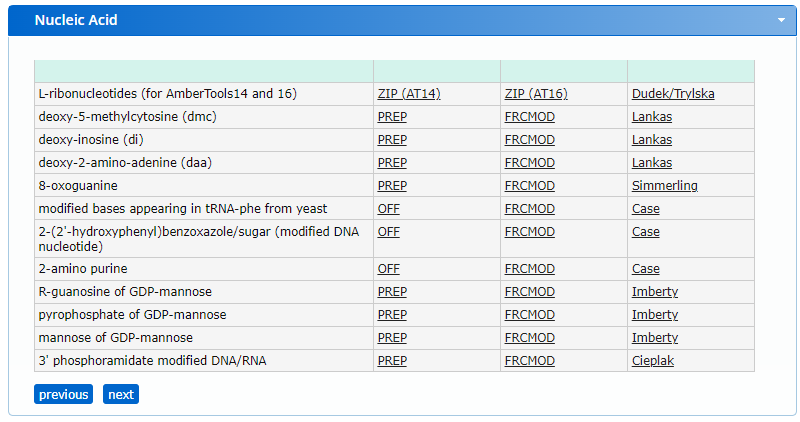
From the website, we find two files, a PREP file and an FRCMOD file. Briefly, the PREP file contains internal coordinate information for non-standard residues, and the FRCMOD (force field modification file) contains additional force field parameters for the non-standard residue. More information of these file formats is available in the AMBER file formats documentation. Using the PREP file, we will generate a PDB structure that we can incorporate in the dna.pdb structure that was generated before.
Ambertools is required to proceed.
Download both PREP and FRCMOD files in the same directory as the dna.pdb structure. Using the LEaP program available in Ambertools, we will generate a PDB structure of the modified residue from the PREP file (the name of the new residue will be DMC). We are going to swap the cytosine #3 of the dna.pdb file with the new residue. Create a text file with the following LEaP commands:
loadamberprep dmc.in savepdb DMC dmc.pdb quit
Save the file as generate-dmc.tleap
The loadamberprep command will read the PREP file dmc.in and load into LEaP a residue with the name DMC (uppercase). The command savepdb will read the structure and information from the DMC variable (that was loaded from the PREP file) and generate a PDB file with the name dmc.pdb. To exit LEaP, the quit command is included.
Run this script with:
tleap -f generate-dmc.tleap
tleap is the text version of LEaP. The -f flag with read our input file.
We now have a dmc.pdb file of the deoxy-5-methylcytosine residue that we can incorporate into the previous dna.pdb file. Download the dmc.pdb file.
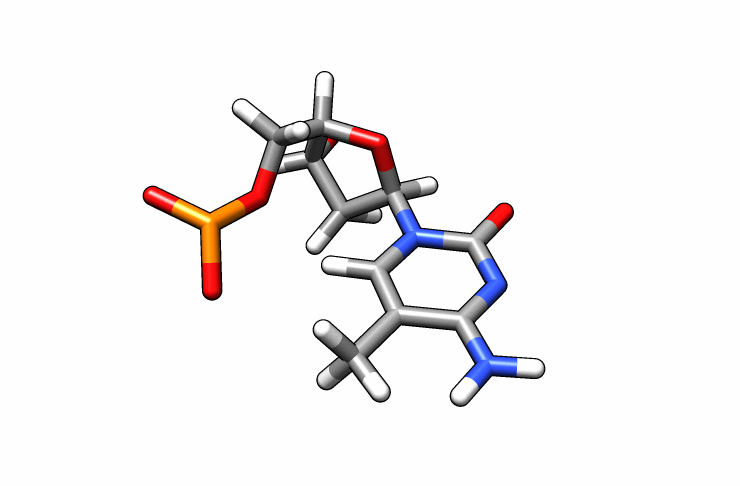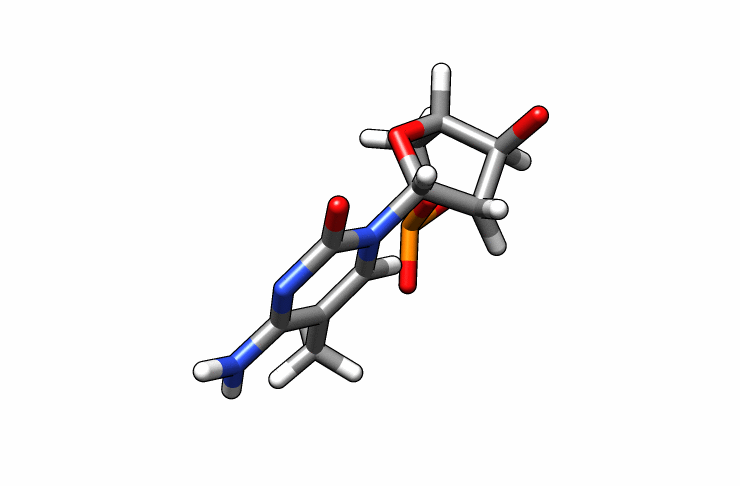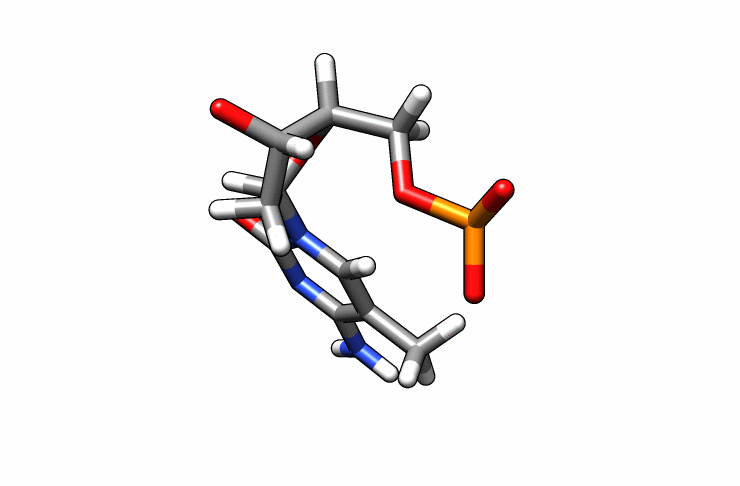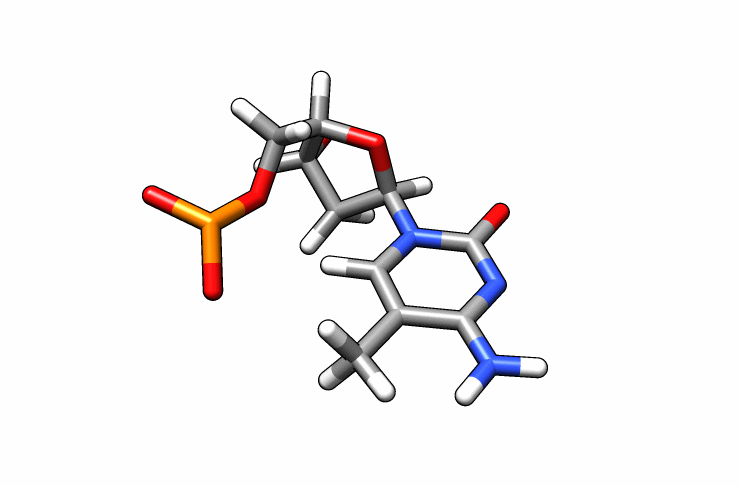
We can now insert our dmc.pdb file into the 3rd position of the dna.pdb file, so we can have the sequence 5′-CG-DMC-GCGCGCG-3′. In a text editor, open the file dna.pdb and look for residue ID #3. Residue ID’s corresponds to column 6 of our dna.pdb file:
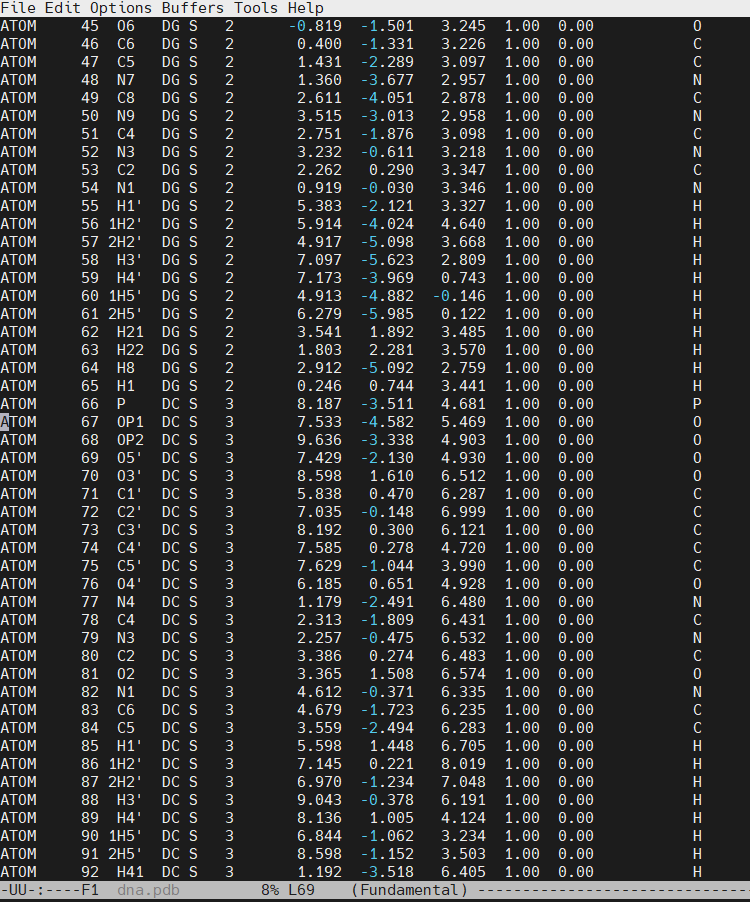
Open the file dna.pdb and delete residue #3, paste the content of the dmc.pdb file. The original cytosine starts at line number 66 in our example.
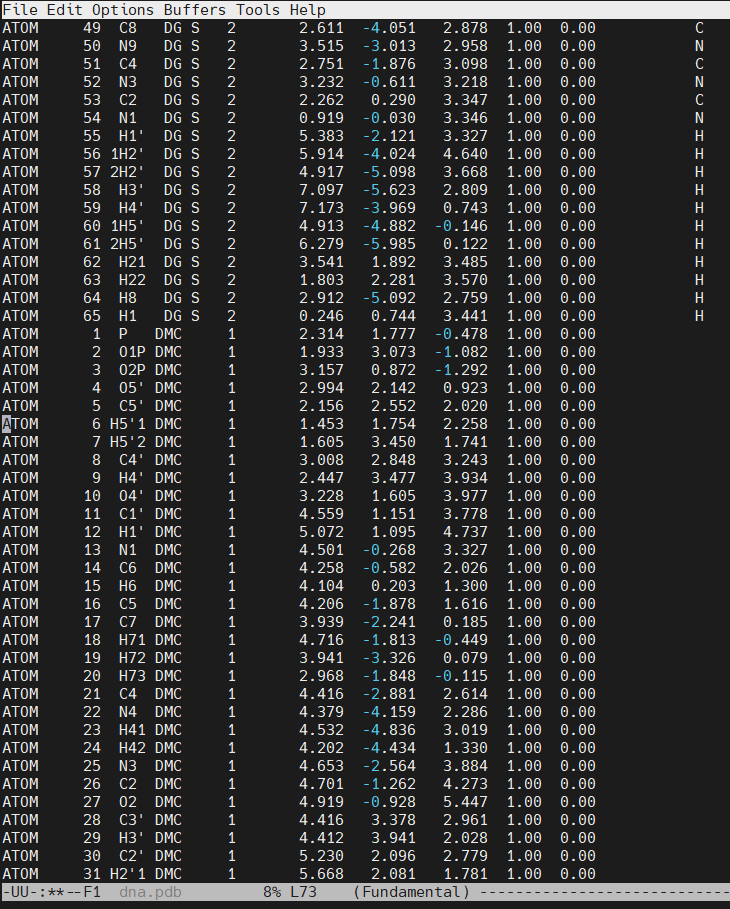
Save the new dna.pdb file that includes the DMC residue (download the dna.pdb file). There will be missing information on some of the PDB file columns. The most important value to update, is the RESIDUE ID NUMBER. At this moment, the DMC residue has a value of 1, this means that we have two residues with the value of 1 (The 5′-cytosine and the DMC). The DMC has to be residue #3 (following the sequence 1C-2G–3DMC–4G-5C-6G-7C-8G-9C-10G). The CPPTRAJ command change will help us fix the residue ID numbers and insert the missing chain name (which in our example is the ‘S’, for sense strand).
Create a text file with the commands:
parm dna.pdb loadcrd dna.pdb name edited change crdset edited oresnums of :1-20 min 1 max 20 change crdset edited chainid to S of :1-10 crdout edited edited.pdb go
Name the file change.cpptraj and run the commands using:
cpptraj -i change.cpptraj
The first change command will edit the resid numbers of residues 1 to 20 starting with 1 and up to resid 20. The second change command will set the chainid to ‘S’ for residues 1 to 10. The output will be a new file called edited.pdb If we open the file in a text editor, we can see the result, the DMC residue now has the chainid ‘S’ and the resid of 3. Download the edited.pdb file.
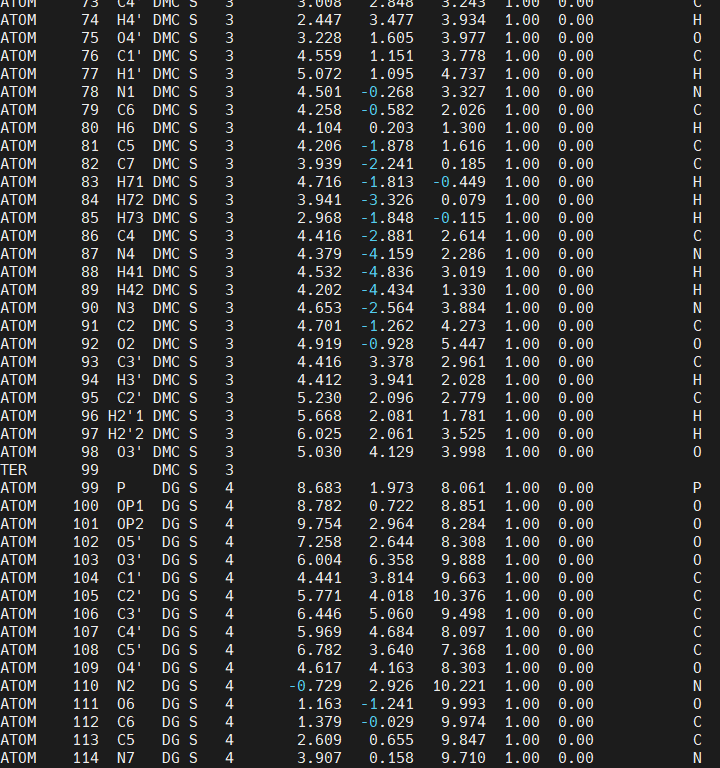
We also notice that there is a TER (termination) card after the DMC residue, it is very important that the TER card is deleted or LEaP will not create the requiered bond between the DMC residue and the DG4 residue when we create the AMBER topology and coordinates file to run simulations.
One last edit we need to perform before simulations can be performed is re-orient the new DMC residue. Open the edited.pdb file in a visualizer and notice how the DMC is overlaping between residues 1 and 2 (highlighted in red outline):
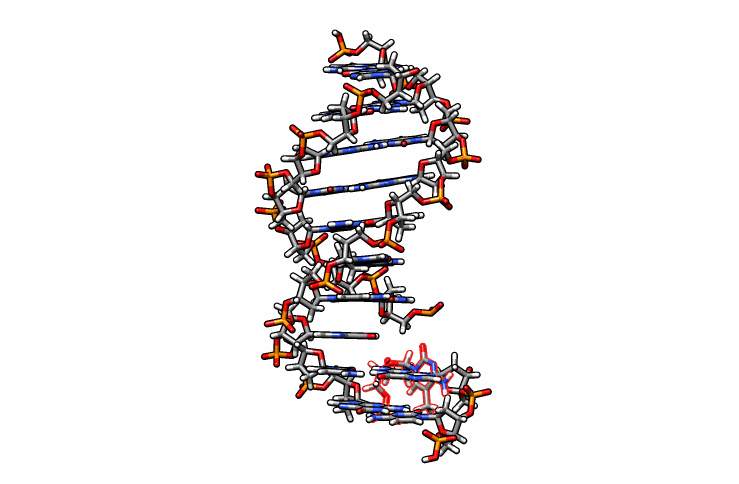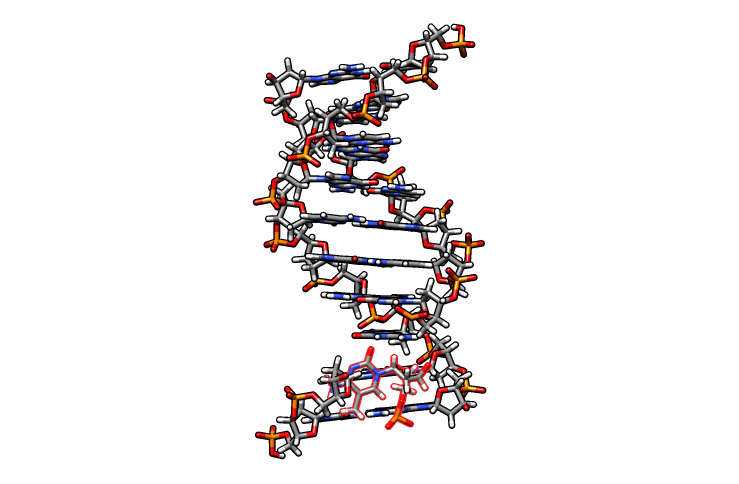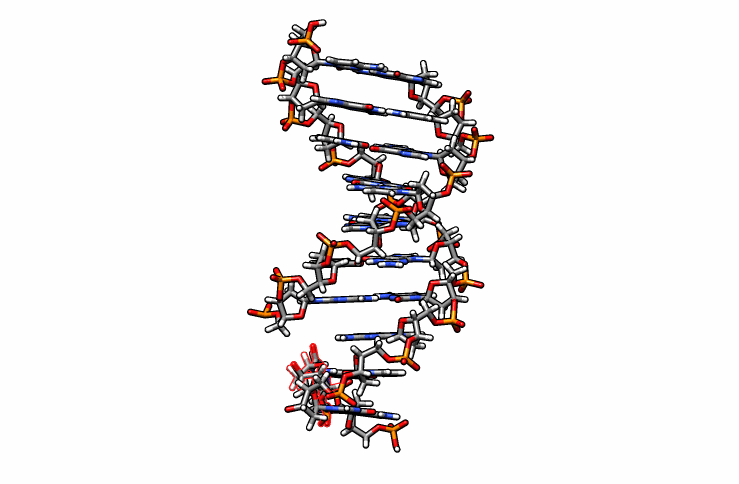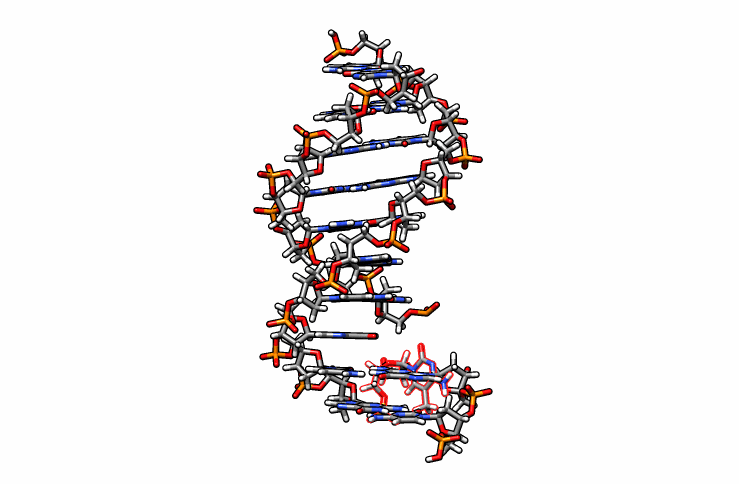
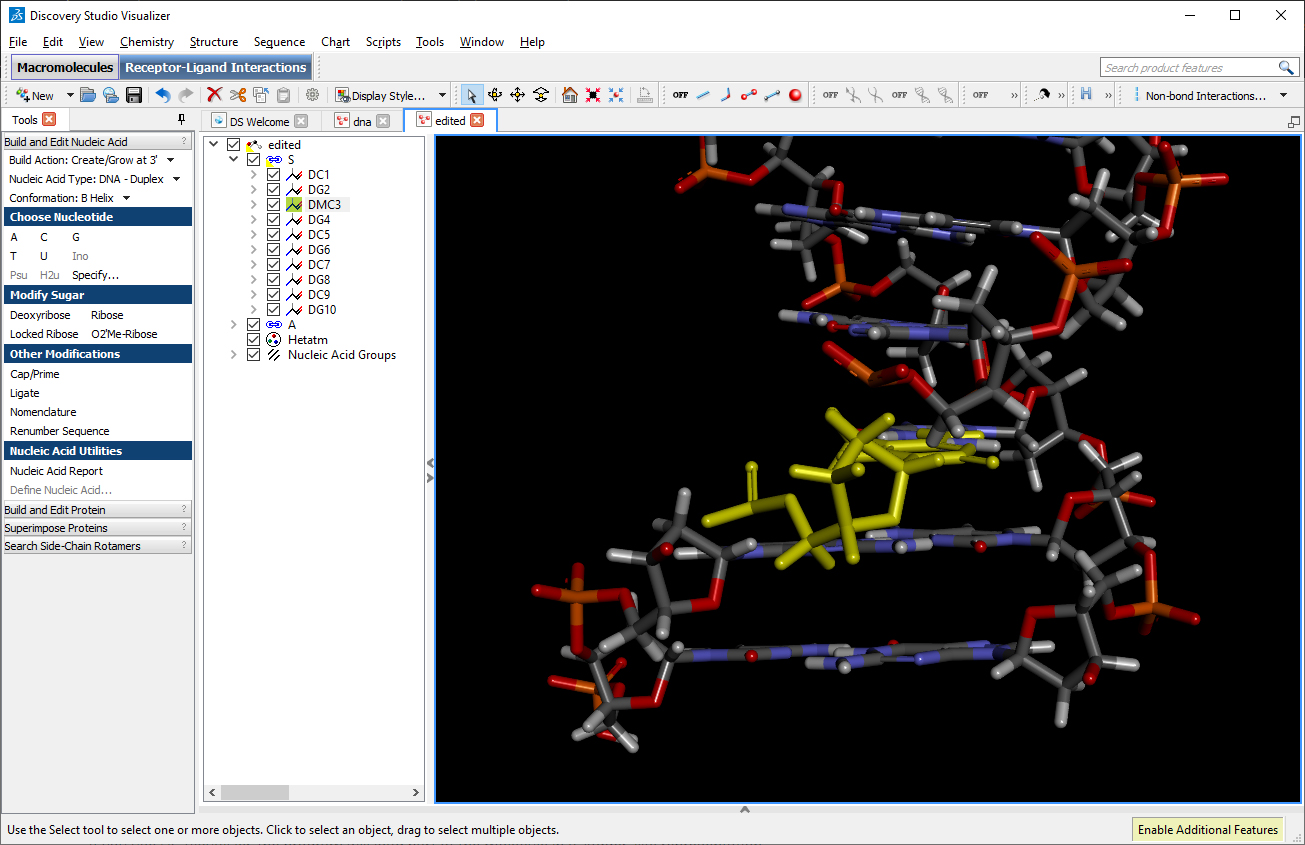
Using DS Visualizer (or any visualization program of choice), manually re-orient the DMC residue until it is in a reasonable position with respect to the backbone and the pairing guanine on the complementary chain (similar to the following image, the reoriented DMC is highlighted in yellow):
The last steps involve making final cleaning of the edited.pdb file before we can use it to generate the required AMBER topology and coordinates files. Sometimes the visualization programs change the name of the hydrogen atoms or add extra atoms that are not recognized by the AMBER force field. To ensure that the edited.pdb file will be correctly processed by LEaP, we first delete the hydrogen atoms with the reduce program (hydrogen atoms will be added by LEaP when we generate the topology). Run the command:
reduce -Trim edited.pdb > edited-noH.pdb
This will generate a new file, edited-noH.pdb without any hydrogen atoms. We can use this file with the following LEaP input file to generate the AMBER topology and coordinates file:
### Generate AMBER topology and coordinates source leaprc.DNA.OL15 source leaprc.water.opc ### Load the DMC library files loadamberprep dmc.in loadamberparams dmc.frcmod x = loadpdb edited-noH.pdb solvateoct x OPCBOX 10.0 addions x Na+ 0 saveamberparm x dna.parm7 dna.crd quit
Save the file as build.tleap and run LEaP using again
tleap -f build.tleap
LEaP (tleap) will run and will output the following unknown atoms:
------- .R<DMC 3>.A<O3' 33> and .R<DG 4>.A<P 1> FATAL: Atom .R<DC5 1>.A<P 29> does not have a type. FATAL: Atom .R<DC5 1>.A<OP1 30> does not have a type. FATAL: Atom .R<DC5 1>.A<OP2 31> does not have a type. FATAL: Atom .R<DC5 1>.A<OP3 32> does not have a type. FATAL: Atom .R<DC5 11>.A<P 29> does not have a type. FATAL: Atom .R<DC5 11>.A<OP1 30> does not have a type. FATAL: Atom .R<DC5 11>.A<OP2 31> does not have a type. FATAL: Atom .R<DC5 11>.A<OP3 32> does not have a type.
Open the edited-noH.pdb file in a text editor and delete the above atoms. LEaP will fill in any required atoms following the force-field templates. After deleting the atoms, we can re-run LEaP and will produce the AMBER topology file dna.parm7 and the coordinates file dna.crd. With these two files we can now run an initial minimization/heat steps to prepare our modifice DNA-duplex for Molecular Dynamics.
We can use the program AmberMdPrep. This wrapper script will prepare explicitly solvated systems for molecular dynamics simulation:
AmberMdPrep.sh -p dna.parm7 -c dna.crd --temp 310