
rms2d | 2drms
Calculate 2D RMSD between frames in 1 or 2 COORDS data sets.
rms2d [crdset <crd set>] [<name>] [<mask>] [out <filename>][dme | nofit | srmsd] [mass][reftraj <traj> [parm <parmname> | parmindex <parm#>] [<refmask>]][corr <corrfilename>]
[crdset <crd set>] Name of previously generated COORDS DataSet. If not specified the default COORDS set will be used.
[<mask>] Mask of atoms to calculate 2D-RMSD for. Default is all atoms.
[out <filename>] Write results to <filename>.
[dme] Calculate distance RMSD instead of coordinate RMSD; this is substantially slower.
[nofit] Calculate RMSD without fitting.
[srmsd] Calculate symmetry-corrected RMSD.
[mass] Mass-weight RMSD.
[reftraj <traj>] Calculate 2D RMSD to frames in trajectory <traj> instead (can also be another COORDS set).
[parm <parmname> | parmindex <#>] Topology to use for <traj>; only useful in conjunction with reftraj.
[<refmask>] Mask of atoms in reference; only useful in conjunction with reftraj.
[corr <corrfilename>] Calculate pseudo-auto-correlation C for 2D-RMSD as
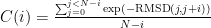
where i is the lag, j is the frame #, and N is the total number of frames. An exponential is used to weight the RMSD since 0.0 RMSD is equivalent to correlation of 1.0. This can only be done if reftraj is not used.
DataSet Aspects:
[Corr] (corr only) Pseudo-auto-correlation.
- Note: For backwards compatibility with ptraj the command ’2drms’ will also work.
A CPPTRAJ recipe for the 2D-RMSD command is available here.
Calculate the best-fit RMSD of each frame in <crd set> (the default COORDS set if none specified) to each other frame. This creates an upper-triangle matrix named <name> (or a full matrix if reftraj specified). The output of the rms2d command can be best-viewed using gnuplot; a gnuplot-formatted file can be produced by giving <filename> a ’.gnu’ extension. For example, to calculate the RMSD of non-hydrogen atoms of each frame in trajectory “test.nc” to each other frame, writing to a gnuplot-viewable file “test.2drms.gnu”:
trajin test.nc rms2d !(@H=) rmsout test.2drms.gnu
To calculate the RMSD of atoms named CA of each frame in trajectory “test.nc” to each frame in “ref.nc” (assuming test.nc and ref.nc are using the default topology file):
trajin test.nc rms2d @CA rmsout test.2drms.gnu reftraj ref.nc