
Creating a hydrogen mass repartitioned topology
You want to create a hydrogen mass repartitioned topology using CPPTRAJ
In order to speed up simulations, a trick that has been used for some time is to use a redistribution of the masses of the atoms. This allows to increase the time between each step in the simulation as reported by Roitberg and collaborators. The main idea is to reduce the mass of heavy atoms that are connected to hydrogen in order to reduce their vibrational speed, which allows for a faster time step in the molecular dynamics engine. Normally only the solute atoms are hydrogen-mass repartitioned and the solute atoms are kept the same. An illustration of doing h-mass repartition for the adenine nucleobase is presented in the next figure:
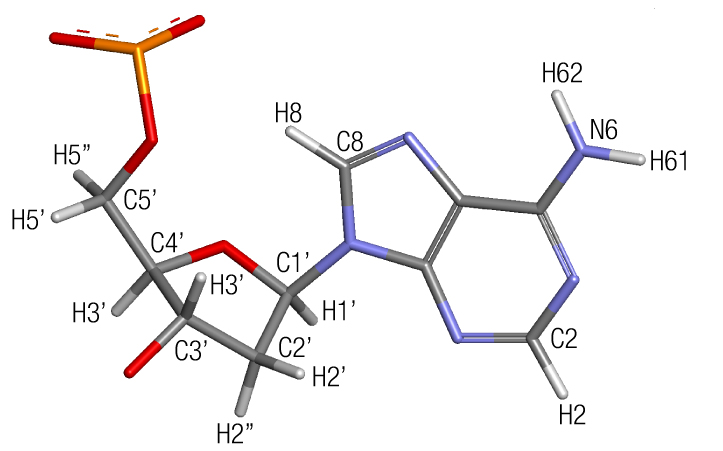
| Atom name | Atom mass before repartition | Atom mass after repartition |
| C5’ | 12.01 | 7.978 |
| H5’ | 1.008 | 3.024 |
| H5’’ | 1.008 | 3.024 |
| C4’ | 12.01 | 9.994 |
| H4’ | 1.008 | 3.024 |
| C3’ | 12.01 | 9.994 |
| H3’ | 1.008 | 3.024 |
| C2’ | 12.01 | 7.978 |
| H2’ | 1.008 | 3.024 |
| H2’’ | 1.008 | 3.024 |
| C1’ | 12.01 | 9.994 |
| H1’ | 1.008 | 3.024 |
| C8 | 12.01 | 9.994 |
| H8 | 1.008 | 3.024 |
| C2 | 12.01 | 7.978 |
| H2 | 1.008 | 3.024 |
| N6 | 14.01 | 9.978 |
| H61 | 1.008 | 3.024 |
| H62 | 1.008 | 3.024 |
From the table, we can see that some mass has been substracted from the heavy atoms and added to the hydrogens. This will allow to run simulations using a higher timestep in the input file:
dt=0.004 ! Time step using H-mass repartition
This increase in time step speeds up the simulation time.
We will use the topology of a 12-mer DNA with the sequence CGCGAATTCGCG as an example (available for download here). In the command line, start CPPTRAJ:
CPPTRAJ: Trajectory Analysis. V6.4.5 ___ ___ ___ ___ | \/ | \/ | \/ | _|_/\_|_/\_|_/\_|_ | Date/time: 04/05/22 15:49:07 | Available memory: 2.341 GB >
Load the topology with the command parm:
CPPTRAJ: Trajectory Analysis. V6.4.5 ___ ___ ___ ___ | \/ | \/ | \/ | _|_/\_|_/\_|_/\_|_ | Date/time: 04/05/22 15:51:09 | Available memory: 2.349 GB > parm dna.prmtop [parm dna.prmtop] Reading 'dna.prmtop' as Amber Topology Radius Set: modified Bondi radii (mbondi) >
Lets display some information about the topology with the command parminfo:
CPPTRAJ: Trajectory Analysis. V6.4.5 ___ ___ ___ ___ | \/ | \/ | \/ | _|_/\_|_/\_|_/\_|_ | Date/time: 04/05/22 15:51:09 | Available memory: 2.349 GB > parm dna.prmtop [parm dna.prmtop] Reading 'dna.prmtop' as Amber Topology Radius Set: modified Bondi radii (mbondi) > parminfo [parminfo] Topology dna.prmtop contains 16074 atoms. Title: default_name Original filename: dna.prmtop 5144 residues. 5122 molecules. 16110 bonds (15566 to H, 544 other). 1476 angles (640 with H, 836 other). 2814 dihedrals (1294 with H, 1520 other). Box: Truncated octahedron 5098 solvent molecules. GB radii set: modified Bondi radii (mbondi) >
This command will print a summary of information contained in the specified topology. We would like to see the difference of a residue when we apply the hydrogen mass repartition command using CPPTRAJ. To get information of a specific residue, we can use the resinfo command:
> resinfo :5 [resinfo :5] 1 residues selected. #Res Name First Last Natom #Orig #Mol C I 5 DA 125 156 32 5 1
In this case we used the flag :5 to tell the resinfo command that we want information for residue #5 (an adenine). For more information about how to select chains, residues or atoms, refer to the atom mask selection syntax page.
To see the mass value of each individual atom in residue #5, we use atominfo:
> atominfo :5 [atominfo :5] 32 atoms selected. #Atom Name #Res Name #Mol Type Charge Mass GBradius El rVDW eVDW 125 P 5 DA 1 P 1.1659 30.9700 1.8500 P 2.1000 0.2000 126 OP1 5 DA 1 O2 -0.7761 16.0000 1.5000 O 1.6612 0.2100 127 OP2 5 DA 1 O2 -0.7761 16.0000 1.5000 O 1.6612 0.2100 128 O5' 5 DA 1 OS -0.4954 16.0000 1.5000 O 1.6837 0.1700 129 C5' 5 DA 1 CJ -0.0069 12.0100 1.7000 C 1.9080 0.1094 130 H5' 5 DA 1 H1 0.0754 1.0080 1.3000 H 1.3870 0.0157 131 H5'' 5 DA 1 H1 0.0754 1.0080 1.3000 H 1.3870 0.0157 132 C4' 5 DA 1 CT 0.1629 12.0100 1.7000 C 1.9080 0.1094 133 H4' 5 DA 1 H1 0.1176 1.0080 1.3000 H 1.3870 0.0157 134 O4' 5 DA 1 OS -0.3691 16.0000 1.5000 O 1.6837 0.1700 135 C1' 5 DA 1 CT 0.0431 12.0100 1.7000 C 1.9080 0.1094 136 H1' 5 DA 1 H2 0.1838 1.0080 1.3000 H 1.2870 0.0157 137 N9 5 DA 1 N* -0.0268 14.0100 1.5500 N 1.8240 0.1700 138 C8 5 DA 1 C2 0.1607 12.0100 1.7000 C 1.9080 0.0860 139 H8 5 DA 1 H5 0.1877 1.0080 1.3000 H 1.3590 0.0150 140 N7 5 DA 1 NB -0.6175 14.0100 1.5500 N 1.8240 0.1700 141 C5 5 DA 1 CB 0.0725 12.0100 1.7000 C 1.9080 0.0860 142 C6 5 DA 1 CA 0.6897 12.0100 1.7000 C 1.9080 0.0860 143 N6 5 DA 1 N2 -0.9123 14.0100 1.5500 N 1.8240 0.1700 144 H61 5 DA 1 H 0.4167 1.0080 1.3000 H 0.6000 0.0157 145 H62 5 DA 1 H 0.4167 1.0080 1.3000 H 0.6000 0.0157 146 N1 5 DA 1 NC -0.7624 14.0100 1.5500 N 1.8240 0.1700 147 C2 5 DA 1 CQ 0.5716 12.0100 1.7000 C 1.9080 0.0860 148 H2 5 DA 1 H5 0.0598 1.0080 1.3000 H 1.3590 0.0150 149 N3 5 DA 1 NC -0.7417 14.0100 1.5500 N 1.8240 0.1700 150 C4 5 DA 1 CB 0.3800 12.0100 1.7000 C 1.9080 0.0860 151 C3' 5 DA 1 C7 0.0713 12.0100 1.7000 C 1.9080 0.1094 152 H3' 5 DA 1 H1 0.0985 1.0080 1.3000 H 1.3870 0.0157 153 C2' 5 DA 1 CT -0.0854 12.0100 1.7000 C 1.9080 0.1094 154 H2' 5 DA 1 HC 0.0718 1.0080 1.3000 H 1.4870 0.0157 155 H2'' 5 DA 1 HC 0.0718 1.0080 1.3000 H 1.4870 0.0157 156 O3' 5 DA 1 OS -0.5232 16.0000 1.5000 O 1.6837 0.1700
This commands shows for residue 5 the atom name, the residue ID (5), the name of the residue (DA), the molecule number of the command (just 1), the type of atom, the charge for each atom, the mass and other properties. Now lets create the hydrogen-mass repartition topology with the hmassrepartition command. Since we only loaded one topology, the hmassrepartition command does not need any additional arguments:
> hmassrepartition [hmassrepartition] Performing hydrogen mask repartitioning for atoms selected by mask '*' Topology is 'dna.prmtop' Hydrogen masses will be changed to 3.024000 amu Skipping solvent. Mask [*] corresponds to 15596 atoms. 272 hydrogen masses modified. 196 heavy atom masses modified.
We can see that the command only adjusted the masses of the solute, leaving the solvent intact. If we run the atominfo command again, we can see that the heavy atoms that are directly bonded with hydrogen, their masses are reduced, and the hydrogen atoms are increased:
> atominfo :5 [atominfo :5] 32 atoms selected. #Atom Name #Res Name #Mol Type Charge Mass GBradius El rVDW eVDW 125 P 5 DA 1 P 1.1659 30.9700 1.8500 P 2.1000 0.2000 126 OP1 5 DA 1 O2 -0.7761 16.0000 1.5000 O 1.6612 0.2100 127 OP2 5 DA 1 O2 -0.7761 16.0000 1.5000 O 1.6612 0.2100 128 O5' 5 DA 1 OS -0.4954 16.0000 1.5000 O 1.6837 0.1700 129 C5' 5 DA 1 CJ -0.0069 7.9780 1.7000 C 1.9080 0.1094 130 H5' 5 DA 1 H1 0.0754 3.0240 1.3000 H 1.3870 0.0157 131 H5'' 5 DA 1 H1 0.0754 3.0240 1.3000 H 1.3870 0.0157 132 C4' 5 DA 1 CT 0.1629 9.9940 1.7000 C 1.9080 0.1094 133 H4' 5 DA 1 H1 0.1176 3.0240 1.3000 H 1.3870 0.0157 134 O4' 5 DA 1 OS -0.3691 16.0000 1.5000 O 1.6837 0.1700 135 C1' 5 DA 1 CT 0.0431 9.9940 1.7000 C 1.9080 0.1094 136 H1' 5 DA 1 H2 0.1838 3.0240 1.3000 H 1.2870 0.0157 137 N9 5 DA 1 N* -0.0268 14.0100 1.5500 N 1.8240 0.1700 138 C8 5 DA 1 C2 0.1607 9.9940 1.7000 C 1.9080 0.0860 139 H8 5 DA 1 H5 0.1877 3.0240 1.3000 H 1.3590 0.0150 140 N7 5 DA 1 NB -0.6175 14.0100 1.5500 N 1.8240 0.1700 141 C5 5 DA 1 CB 0.0725 12.0100 1.7000 C 1.9080 0.0860 142 C6 5 DA 1 CA 0.6897 12.0100 1.7000 C 1.9080 0.0860 143 N6 5 DA 1 N2 -0.9123 9.9780 1.5500 N 1.8240 0.1700 144 H61 5 DA 1 H 0.4167 3.0240 1.3000 H 0.6000 0.0157 145 H62 5 DA 1 H 0.4167 3.0240 1.3000 H 0.6000 0.0157 146 N1 5 DA 1 NC -0.7624 14.0100 1.5500 N 1.8240 0.1700 147 C2 5 DA 1 CQ 0.5716 9.9940 1.7000 C 1.9080 0.0860 148 H2 5 DA 1 H5 0.0598 3.0240 1.3000 H 1.3590 0.0150 149 N3 5 DA 1 NC -0.7417 14.0100 1.5500 N 1.8240 0.1700 150 C4 5 DA 1 CB 0.3800 12.0100 1.7000 C 1.9080 0.0860 151 C3' 5 DA 1 C7 0.0713 9.9940 1.7000 C 1.9080 0.1094 152 H3' 5 DA 1 H1 0.0985 3.0240 1.3000 H 1.3870 0.0157 153 C2' 5 DA 1 CT -0.0854 7.9780 1.7000 C 1.9080 0.1094 154 H2' 5 DA 1 HC 0.0718 3.0240 1.3000 H 1.4870 0.0157 155 H2'' 5 DA 1 HC 0.0718 3.0240 1.3000 H 1.4870 0.0157 156 O3' 5 DA 1 OS -0.5232 16.0000 1.5000 O 1.6837 0.1700
Next, we save the new trajectory with the parmwrite command and the writedata command:
> parmwrite out repartitioned.prmtop [parmwrite out repartitioned.prmtop] Writing topology 0 (dna.prmtop) to 'repartitioned.prmtop' with format Amber Topology Memory used by full exclusion list: 75.872 kB > writedata [writedata]
This new file (repartitioned.prmtop) is now a topology file that has been hydrogen-mass repartitioned and can be used to run simulations using a 4fs timestep.
This process can also be achieved by creating a text file with the name in.cpptraj with the commands:
parm dna.prmtop hmassrepartition parmwrite out repartitioned.prmtop go quit
and running CPPTRAJ with the command:
cpptraj -i in.cpptraj