
Reorienting small molecules
Reorienting small molecules to fit the orientation of another molecule
You want to screen a collection of small molecules into a receptor but need to re-orient them to match a starting structure. In this recipe we will use the graft command to move the coordinates of a small molecule to match the position and orientation of a source molecule so we can do Molecular Dynamics simulation with the new inhibitor.
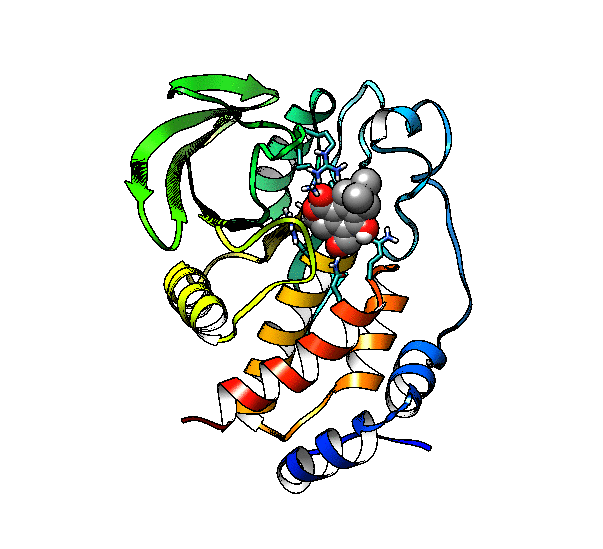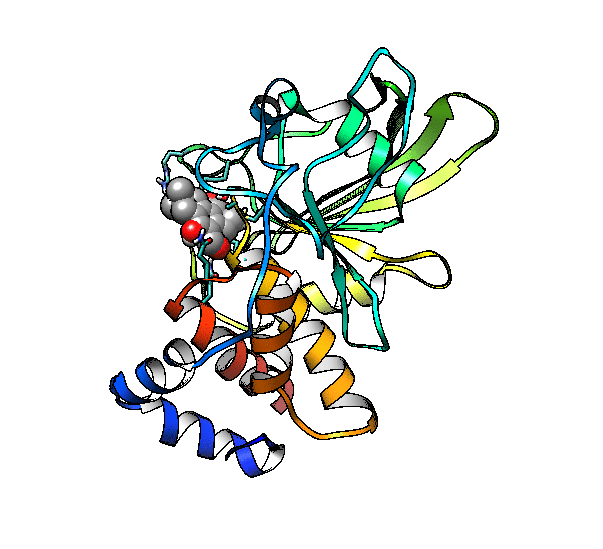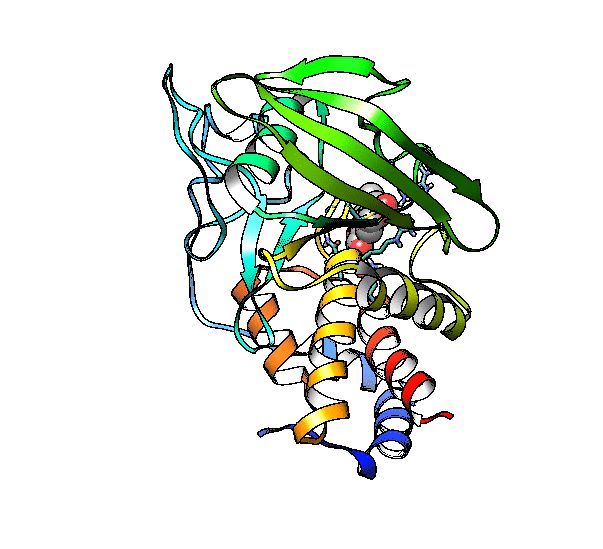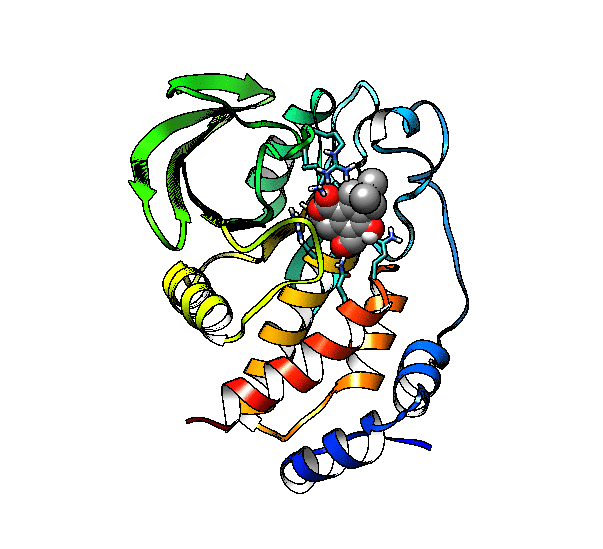
We are going to use the phosphatase 2NV5 with a covalently bound inhibitor molecule in the catalytic site. The idea is to automate the placement of a series of molecules in the same position as the inhibitor so we can test different ligands. For this example, we are going to use our ‘test compound’ and use the graft command to match its position to the illudalic acid that is in the catalytic site so we do not have to manually place it in the same position.
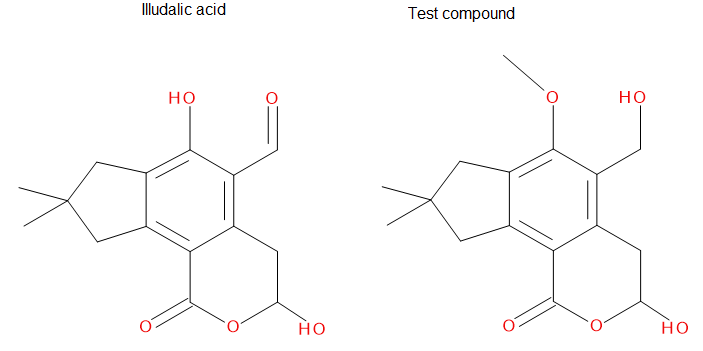
As you can see, the test compound is very similar to the illudalic acid which will facilitate the repositioning. The graft command requires a target and a source to do the RMS fit between the two. The source will be RMS fitted to the target using conventional atom mask syntax. We will also show the use of variables in CPPTRAJ that reduces the amount of commands required.
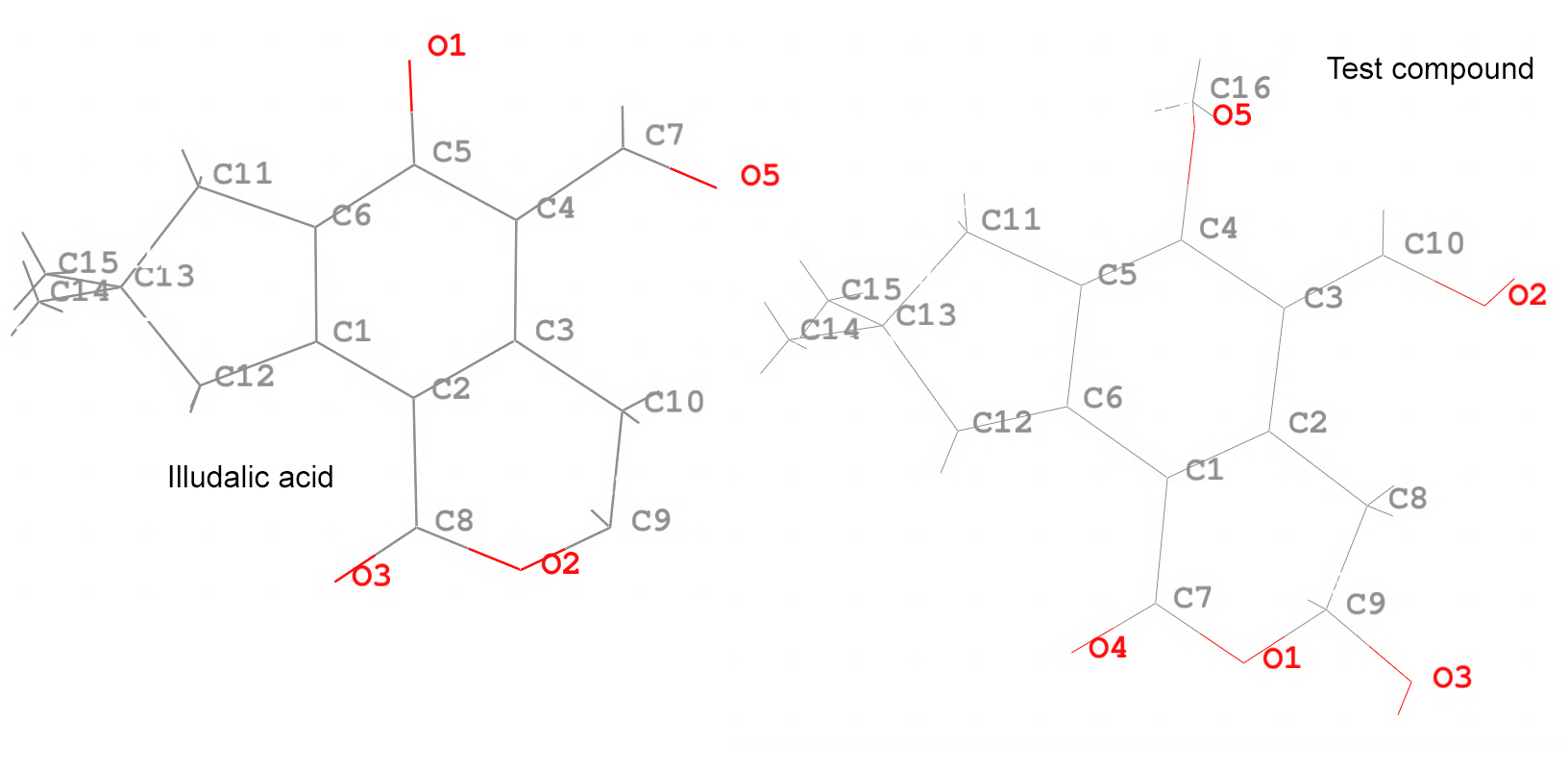
Below is an image of the atom names from each of the molecules, the illudalic acid and the test compound. The graft command will match at least 3 atoms from both molecules, but it is always better to match more atoms to get a better RMS fit.
The CPPTRAJ input file for this look like:
## Load the target set PROTEIN = protein-with-illudalic-acid.pdb ## Set the topology file for the protein parm $PROTEIN ## Load the coordinates of the protein referencing the previous topology loadcrd $PROTEIN parmindex 0 name PROTEIN ## Load the source (which will be matched to the target) set TESTCOMPOUND = test-compound.mol2 ## Load the topology for the test compound parm $TESTCOMPOUND ## Load the coordinates of the test compound referencing the previous topology loadcrd $TESTCOMPOUND parmindex 1 name TESTCOMPOUND ## Call the graft command graft tgt PROTEIN src TESTCOMPOUND name FINAL\ tgtfitmask @O2,C9,C10,C3,C2,C11,C12,C13 srcfitmask @O1,C9,C8,C2,C1,C11,C12,C13 \ tgtmask !(:1-300) srcmask :1 ## Save the compound with new coordinates as a PDB file crdout FINAL test-compound-new-coords.pdb
The graft command requires the target tgt coordinates which are loaded in the PROTEIN set. The test compound src is in the TESTCOMPOUND set. The graft command will create a new data set with the RMS fit structure with the name FINAL. Notice how we use tgtfitmask and srcfitmask to indicate what atom names will be fitted, as it can be seen in the diagrams. The tgtmask of !(:1-300) indicates that we DO NOT want any residues from 1 to 300 in the final structure, this means that all the PROTEIN residues will be deleted from the FINAL structure. For the srcmask, we indicate that we want to include residue #1, which is the only residue that we have.
The crdout command will save the contents of the FINAL dataset, which is the result of our graft command, in the file test-compound-new-coords.pdb.
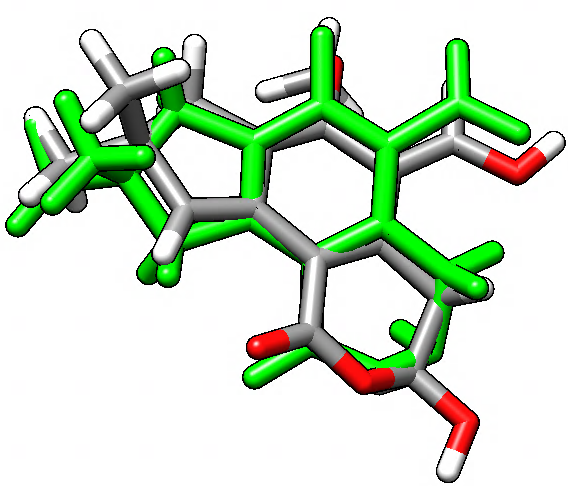
You can see an overlay of the illudalic-acid (green) and the new position of the test compound as produced by the graft command. A better fit could be achieved using more atoms in both tgtfitmask and srcmask. The next step could be to delete the illudalic acid and run MD simulations with this new test compound in the catalytic site of the protein and measure binding energies and potential changes in the structure and dynamics of the complex.
It is important to mention that the graft command provides extra functionality than just a plain RMS fit, which includes the deleting of atoms and the creation of bonds between selected atoms or residues.