
radial | rdf
Calculate radial distribution function.
radial [out <outfilename>] <spacing> <maximum> <solvent mask1> [<solute mask2>]
[noimage] [mass]
[density <density> | volume] [<dataset name>] [intrdf <file>] [rawrdf <file>]
[{{center1|center2|nointramol|toxyz <x>,<y>,<z>} |
[byres1] [byres2] [bymol1] [bymol2]}]
out <outfilename> File to write RDF to, required.
<spacing> Bin spacing, required.
<maximum> Max bin value, required.
<solvent mask1> Atoms to calculate RDF for, required.
[<solute mask2>] (Optional) If specified calculate RDF of all atoms in <solvent mask1> to each atom in <solute mask2>.
[noimage] Do not image distances.
[mass] Use center of mass for centerX/byresX/bymolX keywords, otherwise use geometric center
[density <density>] Use density value of <density> for normalization (default 0.033456 molecules 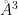
[volume] Determine density for normalization from average volume of input frames.
<dataset name> Name of output data sets.
[intrdf <file>] Calculate integral of RDF bin values (averaged over # of frames but otherwise not normalized) and write to <file> (can be same as <output_filename>).
[rawrdf <file>] Write raw (non-normalized) RDF values to <file>.
[center1] Calculate RDF from center of atoms in <solvent mask1> to all atoms in <solute mask2>.
[center2] Calculate RDF from center of atoms in <solute mask2> to all atoms in <solvent mask1>.
[nointramol] Ignore intra-molecular distances.
[toxyz <x>,<y>,<z>] Calculate RDF from atoms in <solvent mask1> to point specified by <x> <y> and <z> (in Ang.).
[byres1] Calculate using the centers of each residue in the first mask.
[bymol1] Calculate using the centers of each molecule in the first mask.
[byres2] Calculate using the centers of each residue in the second mask.
[bymol2] Calculate using the centers of each molecule in the second mask.
DataSet Aspects:
<setname> The radial distribution function.
<setname>[int] (intrdf only) Integral of RDF bin values.
<setname>[raw] (rawrdf only) Raw (non-normalized) RDF values.
Calculate the radial distribution function (RDF, aka pair correlation function) of atoms in <solvent mask1> (note that this mask does not need to be solvent, but this nomenclature is used for clarity). If an optional second mask (<solute mask2>) is given, calculate the RDF of ALL atoms in <solvent mask1> to EACH atom in <solute mask2>. If desired, the geometric center of atoms in <solvent mask1> or <solute mask2> can be used by specifying the center1 or center2 keywords respectively, or alternatively intra-molecular distances can be ignored by specifying the nointramol keyword. If byresX or bymolX are specified, distances will be between the centers of residues/molecules selected by <solvent mask1> or <solute mask2>. If mass is specified use the centers of mass for centerX/byresX/bymolX, otherwise use geometric center.
The RDF is calculated from the histogram of the number of particles found as a function of distance 
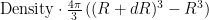
where 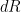
The default density value is 0.033456 molecules 

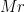
volume keyword is specified the density is determined from the average volume of the system over all Frames.
Note that correct normalization of the RDF depends on the number of atoms in each mask; if multiple topology les are being processed that result in changes in the number of atoms in each mask, the normalization will be off.
The basic (i.e. no center1/center2/byres1/byres2/bymol1/bymol2) RDF calculations are now CUDA parallelized. However, the calculation is done in single precision on GPUs so the resulting histograms may differ slightly from the CPU (on the order of 0.0002- 0.0004).