
lie
Calculate linear interaction energy between user-specified ligand and surroundings.
lie [<name>] <Ligand mask> [<Surroundings mask>] [out <filename>] [nopbc] [noelec] [novdw] [cutvdw <cutoff>] [cutelec <cutoff>] [diel <dielc>]
DataSet Aspects:
[EELEC] Electrostatic energy (kcal/mol).
[EVDW] van der Waals energy (kcal/mol).
For each frame, calculate the non-bonded interactions between all atoms in <Ligand mask> with all atoms in <Surroundings mask>. Electrostatic and van der Waals interactions will be calculated for all atom pairs. A separate electrostatic and van der Waals cutoff can be applied, the default is 12.0 Angstroms for both. <dielc> is an optional dielectric constant. Either the electrostatic or van der Waals calculations can be suppressed via the keywords noelec and novdw, respectively.
Periodic boundary conditions (and the minimum image convention) can be abandoned with the nopbc keyword. Note, however, that no prior imaging is performed if the frames contain periodic boundaries. This may be useful for instances when you are simulating a microscopic droplets. The electrostatic interactions are calculated according to a simple shifting function shown below.
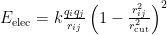
The data file will contain two data sets—one for electrostatic interactions and one for van der Waals interactions. Periodic topologies and trajectories are required (i.e., explicit solvent is necessary). The minimum image convention is followed.