
Hydrogen bond analysis within a protein
You want to study the hydrogen-bonds within a protein system
This recipe focuses on the analysis of intra-molecular hydrogen bonds as a protein unfolds at high temperature. For an example about hydrogen bonds between two independent systems (i. e. between a protein and a small molecule) follow this recipe.
Hydrogen bonds are an important non-covalent structural force (primarily electrostatic in nature) in molecular systems. They are formed when a single hydrogen atom is effectively shared between the heavy atom it is covalently bonded to (the hydrogen bond donor) and another heavy atom (the hydrogen bond acceptor). Both the donor and acceptor atoms are typically quite electronegative.
The general rule of thumb for determining whether atoms can hydrogen bond is “hydrogen bonds are FON”, i.e. hydrogens bonded to F, O, and N atoms can be donated, and F, O, and N atoms can be acceptors (although there are exceptions).
CPPTRAJ will find and track hydrogen bonds over the course of a trajectory. Hydrogen bonds are determined using simple geometric criteria: the donor to acceptor heavy atom distance, and optionally the donor-hydrogen-acceptor angle. Hydrogen bond donors and acceptors can either be searched for automatically (using the FON criterion) or can be explicitly defined by the user via atom masks. CPPTRAJ will also track solute to solvent hydrogen bonds and solvent bridging interactions (i.e. two or more solute residues hydrogen bonded to a single solvent residue).
Note that in nature, the strength of a hydrogen bond is determined by both distance and angle (having to do with optimal overlap of molecular orbitals). However in most pairwise additive force fields (e.g. Amber, Charmm) hydrogen bond interactions arise solely from non-bonded terms and therefore their strength is not dependent on angle.
In this example we will use CPPTRAJ to find and track hydrogen bonds during the unfolding of the model beta hairpin peptide Trpzip2 (PDB 1LE1) at 345 K.
- trpzip2.ff10.tip3p.parm7: Trpzip2 solvated topology file.
- trpzip2.unfold.nc: MD trajectory, unfolding of Trpzip2 at 345 K.
- cpptraj.hbond.in: CPPTRAJ input.
Note that although input is provided in a file, users are encouraged to use the interactive mode to become better familiar with CPPTRAJ workflow and command options.
Performing Hydrogen Bond Analysis with CPPTRAJ
Start CPPTRAJ by typing ‘cpptraj’. Load the topology and trajectory with the following commands:
parm trpzip2.ff10.tip3p.parm7 trajin trpzip2.unfold.nc
The first hbond command will be used to track all solute-solute and solute-solvent hydrogen bonds (all solvent residues are named WAT, the Amber default), as well as solute-solvent-solute bridging interactions:
hbond All out All.hbvtime.dat solventdonor :WAT solventacceptor :WAT@O \ avgout All.UU.avg.dat solvout All.UV.avg.dat bridgeout All.bridge.avg.dat
The number of solute-solute and solute-solvent hydrogen bonds, as well as the number of bridges and the identity of bridging residues will be written to ‘All.hbvtime.dat’. Average statistics on each found hydrogen bond will be found in the files ‘All.UU.avg.dat’, ‘All.UV.avg.dat’, and ‘All.bridge.avg.dat’.
The second hbond command will be used to track only solute-solute backbone hydrogen bonds. As such we will provide a mask so that only C, O, N, and H atoms will be considered as potential donors/acceptors. We will also specify the series keyword so that the time series of each found hydrogen bond is recorded for further analysis (1 for hydrogen bond present, 0 for not present).
hbond Backbone :1-12@C,O,N,H avgout BB.avg.dat series uuseries bbhbond.gnu
The uuseries keyword will write the raw hydrogen bond time series data to bbhbond.gnu, which can be visualized using gnuplot.
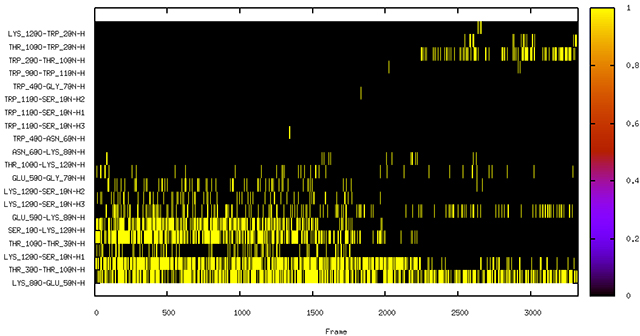
We will now issue a command that will create a file containing only the number of solute-solute hydrogen bonds (aspect: [UU]) from both commands, as well as
number of solute-solvent hydrogen bonds (aspect: [UV]) and number of solute-solvent-solute bridges (aspect: [Bridge]).
create nhbvtime.agr All[UU] Backbone[UU] All[UV] All[Bridge]
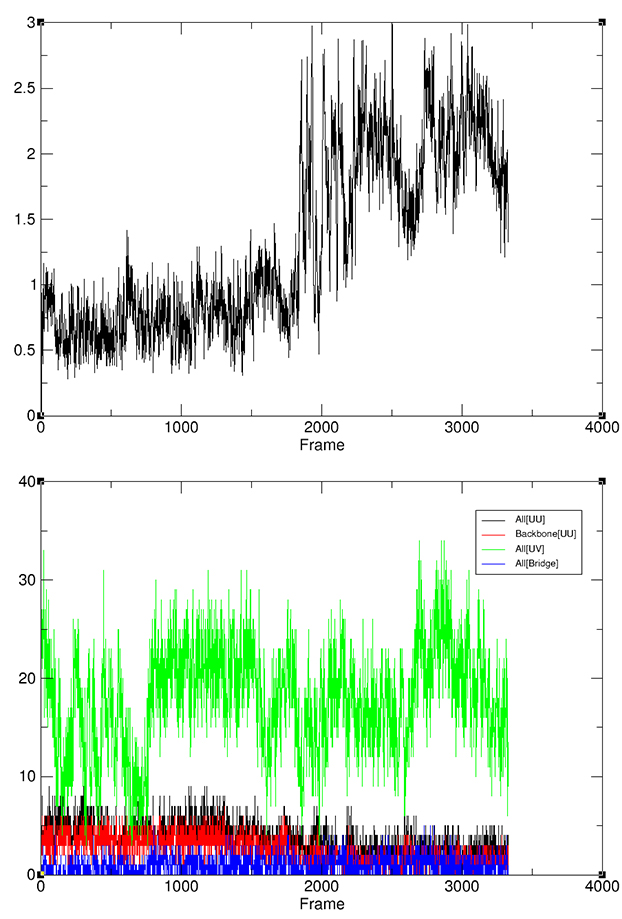
Finally, we will add an RMSD command to calculate backbone RMSD to the first frame as a means to track unfolding (Note this is not part of hbond analysis):
rms BBrmsd :1-12@C,CA,N out BBrmsd.agr
Note that at this point although we have set up hydrogen bond commands, there isn’t actually any hydrogen bond data yet. That is because CPPTRAJ needs to actually process trajectories before it can find the hydrogen bonds. So now issue a run command to actually find the hydrogen bonds.
When the run has completed take some time to review the output files. In particular note the point at which the backbone RMSD increases (BBrmsd.agr, top plot), and see how that change correlates with changes in the number of solute-solute and solute-solvent hydrogen bonds (nhbvtime.agr, bottom plot).
Once the run has completed and hydrogen bonds have been found, we can perform subsequent analysis on the hydrogen bond time series. The following commands will perform lifetime analysis on the backbone hydrogen bonds:
lifetime Backbone[solutehb] out backbone.lifetime.dat runanalysis
This will also generate so-called lifetime curves in ‘crv.backbone.lifetime.dat’, which is a means of visualizing the time-dependent behavior of the hydrogen bonds.
Another example of the Hbond command is available in this recipe