
Create a new nucleotide
Create a new AMBER-compatible nucleotide to perform MD simulations.
In this recipe, we will use CPPTRAJ (and several other programs) to create a new nucleotide that is compatible with AMBER to run Molecular Dynamics simulations. Please notice that there are multiple ways, programs and methods to generate a new residue, the purpose of this recipe is to show how to integrate CPPTRAJ functionalilty.
We will use as an example a thymine derivate on which the pentofuranose ring of DNA is replaced with a seven-membered (oxepane) sugar ring (see the work of Damha and collaborators for an example). We are going to build the structure and required parameters to create our new residue (example name: O37) that will have a 3′-7′ linkage (instead of the 5′-7′ linkage from the Damha article) within a DNA chain.
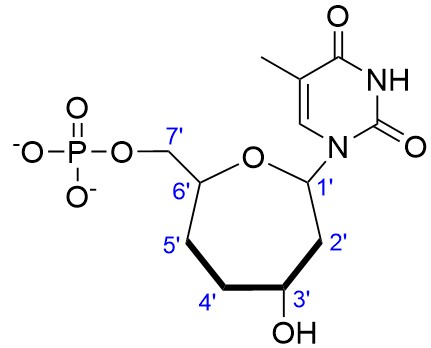
As a starting point, we will use the deoxythymine molecule as generated directly from LEaP, this helps to retain the same atom names of the base. In tleap, run:
user@computer$ tleap source leaprc.DNA.OL21 savepdb DT DT.pdb quit
We have now our DT.pdb file that we can use as a starting point to create our oxepane analog. Using a molecular graphics software, we alter the 5-member ring into our 7-member ring:
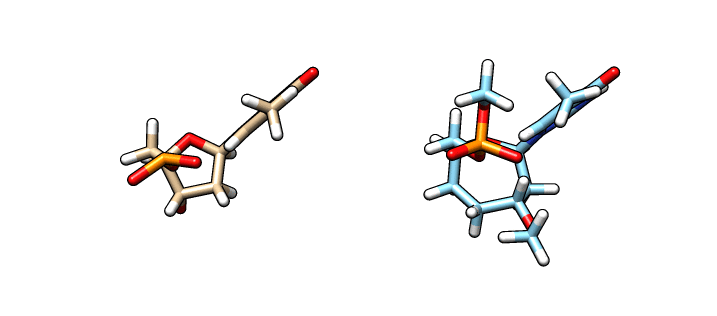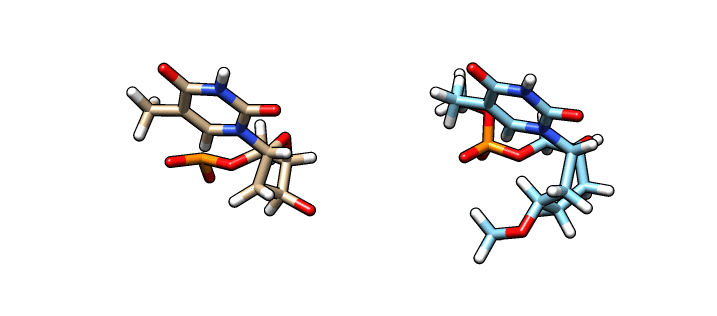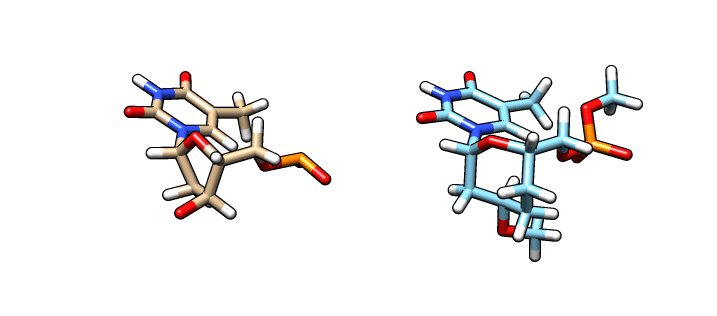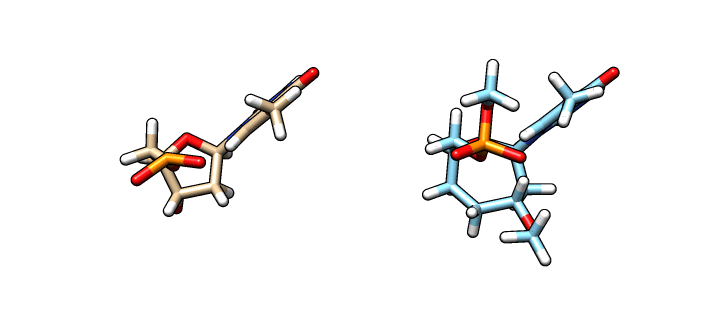
In the left is the DT.pdb file we created from LEaP and in the right (blue) is the modified ‘O37′ residue – we are going to name this file o37-for-charges.pdb. In order to calculate the charges we need to add capping groups in our new residue. Notice the methyl capping group in the 3’-cargon and the oxy-methyl capping group in the phosphate group. To calculate the charges we are going to use antechamber (from AmberTools) to generate an input file for Gaussian:
antechamber -i o37-for-charges.pdb -fi pdb -o o37-charges.com -fo gcrt -nc -1
Notice the -nc flag set to -1 to indicate the overall charge of the residue. This will generate an input file for Gaussian with the correct internal option keywords (iop) to calculate the charges for our new residue. When this calculation is done, we will have a o37-charges.log file. We now can use this file to refit the charges following the RESP methodology using antechamber again:
antechamber -i o37-charges.log -fi gout -o o37-with-charges.mol2 -fo mol2 -rn O36 -at AMBER -c resp
This will read the charges from the Gaussian output, fit the charges using the RESP methodology, rename the residue to O37, assign atom types using AMBER atom types and generate a o37-with-charges.mol2 file. Because Gaussian does not use atom types, we have lost the atom names from our original template (o37-for-charges.pdb). We can use CPPTRAJ to map the names of the atoms that we have in the o37-for-charges.pdb file to our new o37-with-charges.mol2 file. For that, we use the atommap keyword and the following CPPTRAJ script:
parm o37-with-charges.mol2 reference o37-with-charges.mol2 parm o37-for-charges.pdb reference o37-for-charges.pdb parmindex 1 atommap o37-with-charges.mol2 o37-for-charges.pdb mapout atommap.dat changenames trajin o37-with-charges.mol2 trajout o37-with-charges-reordered.mol2 mol2
This new file: o37-with-charges-reordered.mol2 now has the charges, atom types and the atom names from our original starting structure. We now need to remove the capping groups marked in yellow:
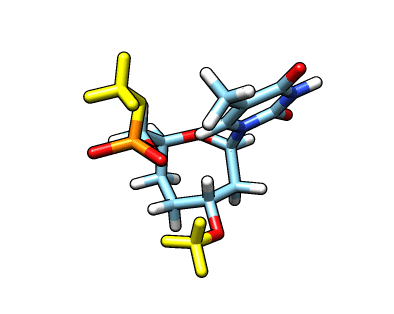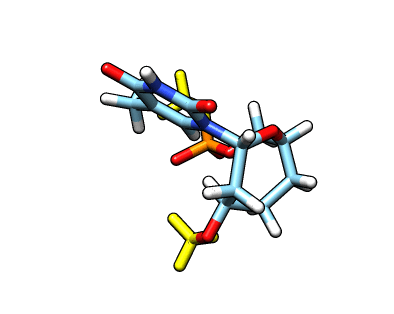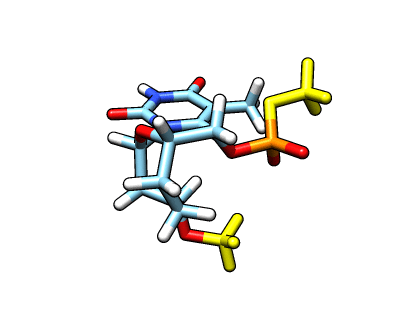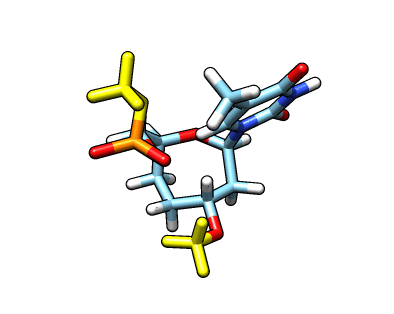
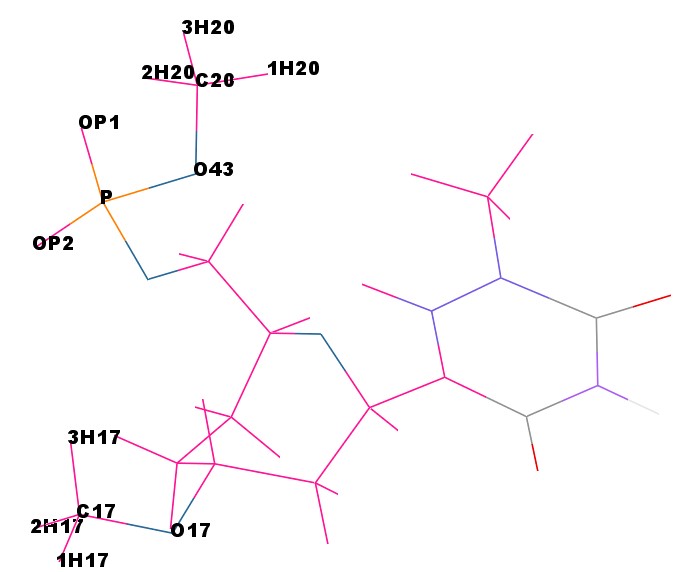 The atom names for the cap at the phosphate are
The atom names for the cap at the phosphate are @O43,C20,1H20,2H20,3H20 and the atom names for the cap in the C3′ position are @C17,1H17,2H17,3H17. We can use CPPTRAJ strip command to remove the capping groups and to refit the charge to the integer value of -1:
parm o37-with-charges-reordered.mol2 name sugar trajin o37-with-charges-reordered.mol2 parm sugar strip @O43,C20,1H20,2H20,3H20 strip @C17,1H17,2H17,3H17 charge -1.0 trajout o37-fragment.mol2 mol2 run
This will generate a new file called o37-fragment.mol2 without the capping groups, a total charge of -1 and AMBER atom names. We can now generate a library file that we can use to create AMBER topology and coordinate files. With this tleap script we load the o37-fragment.mol2, assing head and tail atoms and generate a library file:
source leaprc.DNA.OL21 O37 = loadmol2 o37-fragment.mol2 set O37 restype nucleic set O37 name O37 set O37 head O37.1.P set O37 tail O37.1.O17 saveoff O37 O37.lib quit
This script can be saved in a text file (for example – generate-lib.tleap) and run in this manner:
tleap -s -f generate-lib.tleap
The script will load the o37-fragment.mol2 file, set the residue type to nucleic, the name of the residue to O37, the head of the residue to the atom P and the tail to the atom O17. Finally it will create an AMBER library file with the name O37.lib. This library file can now be used in a simulation – for example – we can use the crystal structure of the DNA dodecamer (sequence CCGCGAATTCGCG) and insert our new residue. First we download the 1bna.pdb file from the PDB. To clean the PDB, we can use the prepareforleap command using the following CPPTRAJ script:
parm 1bna.pdb loadcrd 1bna.pdb name edited prepareforleap crdset edited name from-prepareforleap \ out from-prepareforleap-tleap.in leapunitname x \ pdbout from-prepareforleap.pdb nowat noh go
A way to change one residue to another, when some of the atom names are similar, is to change the residue name directly into the PDB file, delete the atoms that are not involved in the new residue and let LEaP to fill the missing atoms. To do this for this example, we open the DNA file from-prepareforleap.pdb created with the previous CPPTRAJ command in a text editor and change deoxythymine #7 from DT to O37 (which is the name of our new residue in the library file) – the original file is:
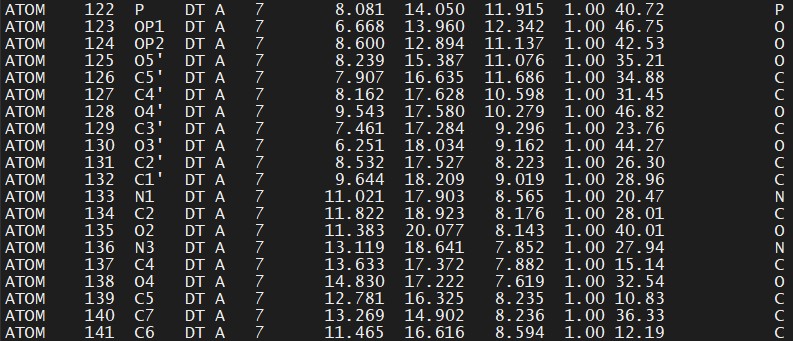
We substitue DT for O37 (delete the line with the O3′ atom, since in our example it has another name. This will be fixed by LEaP):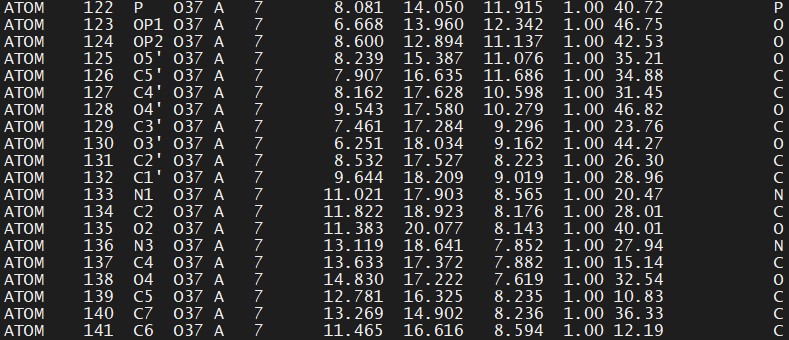
With this PDB file, we can now use LEaP to add water molecules, counter ions, and generate the AMBER topology and coordinates:
###################################### source leaprc.DNA.OL21 source leaprc.water.opc loadoff O37.lib ###################################### x = loadpdb from-prepareforleap.pdb solvateoct x OPCBOX 10.0 addions x MG 8 addions x Na+ 0 addions x Cl- 0 ## Extra ions addions x Na+ 40 addions x Cl- 40 saveamberparm x complex.parm7 complex.coords quit
An example simulation is presented below: