Compare the surface area of two cyclic peptides
You want to calculate how a specific change in the structure of a cyclic peptide alters the surface area.
For this recipe, we are going to use three bicycle peptides that have been reported by Bennett and colaborators in the publication: Identification and Optimization of EphA2-Selective Bicycles for the Delivery of Cytotoxic Payloads (link). In this work, they synthesize a series of peptides that are joined together by a scaffold molecule, forming two loops, hence the name bicycle peptides:
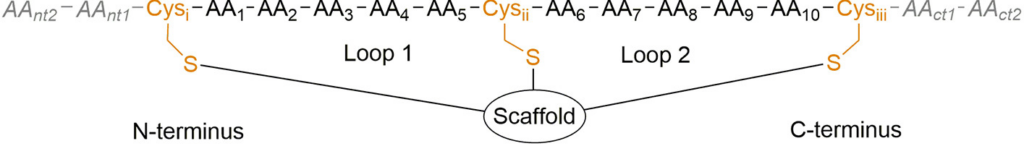
For our recipe, we are going to use the bicycle peptide that they have reported in the crystal structure PDB ID: 6RW2 and perform molecular dynamics simulations with three of the scaffold molecules presented below (from the article):
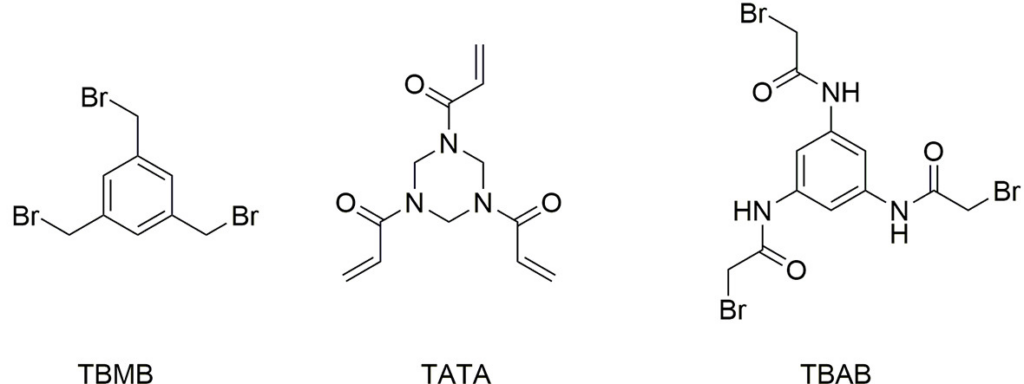
Each of those scaffolds will be included in three different simulations and we will analyse the results. The systems were built using the AMBER force field ff19SB and calculated for 1 micro second with three independent replicas. A snapshot of the resulting simulations is presented in the video below:
The peptide sequence is: Ala-Arg-Asp-Cys-Pro-Leu-Val-Asn-Pro-Leu-Cys-Leu-His-Pro-Gly-Trp-Thr-Cys and the scaffold at the end, linking the three cysteines to form the bicycle configuration. The total amount of residues is 19. We will use the CPPTRAJ comand surf to calculate the surface area in 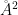
parm nowater.parm7 trajin replica1/nowater.nc 1 last 1 trajin replica2/nowater.nc 1 last 1 trajin replica3/nowater.nc 1 last 1 surf R1 :1-19 out surf.dat
The parm command will read the topology, the trajin command will read each of the pre-processed trajectories, each representing an independent simulation. The ‘1 last 1’ keywords in the trajin command reads as: start in frame #1, read until the last frame and read every 1th frame. Note that there is also a command named molsurf, which extracts the atoms in the mask and calculates the surface area of those extracted atoms, completely ignoring any other atoms that might be present in the solute. In our case, the use of surf will compute the summed contributions of the atoms in the mask to the total surface area of the solute.
We run the same CPPTRAJ input script for each of the systems. The surf.dat file will look like:
#Frame R1 1 1809.9171 2 1721.8772 3 1807.1048 4 1661.1966 5 1701.9476 6 1877.4260 7 1914.3335 8 1757.9907 9 1695.7402
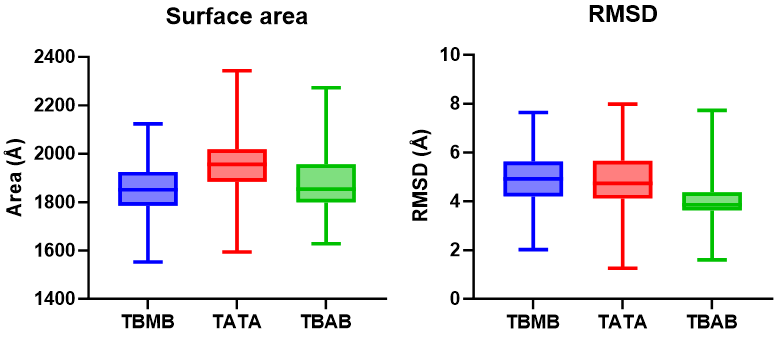
The R1 value will correspond to the measured surface area for each frame. Plotting each of the results we get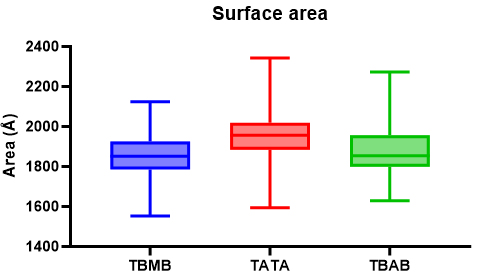
The analysis suggest that if we change the scaffold molecule for TBMB and TBAB, the overall surface area is reduced. We can extend the analysis if we calculate the root mean square deviation of each of the systems over time with the rms command:
parm nowater.parm7 trajin replica1/nowater.nc 1 last 1 trajin replica2/nowater.nc 1 last 1 trajin replica3/nowater.nc 1 last 1 rms :1-19 out rms.dat surf R1 :1-19 out surf.dat
We can now plot the information from the surf command and the rms command side by side to study the results of the simulations: