
atomicfluct | rmsf
Compute the atomic positional fluctuations.
atomicfluct [<name>] [out <filename>] [<mask>] [bfactor] [calcadp [adpout <file>]] [{byres [pdbres] [resrange <range>] | byatom | bymask}] [start <start>] [stop <stop>] [offset <offset>]
<name> Output data set name.
out < filename> Write data to file named <filename>
[<mask>] Calculate fluctuations for atoms in <mask> (all if not specified).
[bfactor] Calculate atomic positional fluctuations squared and weight by 8/3π^2; this is similar but not necessarily equivalent to the calculation of crystallographic B-factors.
[calcadp [adpout < file>]] Calculate anisotropic displacement parameters and optionally output them to <file>.
byres [pdbres] Output the average (mass-weighted) fluctuation by residue.
pdbres If specified, the original residue numbering will be used.
resrange <range> Calculate fluctuations for residues in the given range argument. The residues selected by <mask> must be a subset of residues specified with <range>. Unselected residues will get a fluctuation of 0.0.
bymask Output the average (mass-weighted) fluctuation for all atoms in <mask>.
byatom (default) Output the fluctuation by atom.
[<start>] Frame to begin calculation at (default 1).
[<stop>] Frame to end calculation at (default last).
[<offset>] Frames to skip between calculations (default 1).
Data sets created
<name> Hold atomic fluctuations.
<name>[ADP] Hold anisotropic displacemente parameters if calcadp specified.
Compute the atomic positional fuctuations (also referred to as root-mean-square fuctuations, RMSF) for atoms specifed in the mask. The RMSF of a given atom i is calculated as:

where 
Note that RMS fitting is not done implicitly. If you want fluctuations without rotations or translations (for example to the average structure), perform an RMS fit to the average structure (best) or the first structure (see rmsd) prior to this calculation.
The units are 
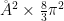
bfactor is specified.
If byres or bymask are specified, the mass-weighted average of atomic fluctuations of each atom for either each residue or the entire mask will be calculated respectively:

If calcadp is specified, anisotropic displacement factors for atoms will be calculated and written to the file specified by adpout (or STDOUT if not specified) using PDB ANISOU record format. Note that calcadp automatically implies bfactor.
With CPPTRAJ it is possible to perform coordinate averaging, the fit to average coordinates, and the atomic fluctuation calculation in a single execution like so:
parm myparm.parm7 trajin mytrajectory.crd rms first average crdset MyAvg run rms ref MyAvg atomicfluct out fluct.agr
To write the mass-weighted B-factors for the protein backbone atoms C, CA, and N, averaged by residue use the command:
atomicfluct out back.agr @C,CA,N byres bfactor
To write the RMSF or atomic positional fluctuations of the same atoms, use the command:
atomicfluct out backbone-atoms.agr @C,CA,N
To write a PDB of averaged coordinates (after fitting to the first frame) with both B-factors and anisotropic temperature factors:
parm myparm.parm7 trajin mytraj.nc rms first average crdset MyAvg atomicfluct MyFluct calcadp run crdout MyAvg mypdb.pdb adpdata MyFluct[ADP] bfacdata MyFluct