
Measuring the RMSD of a protein system.
This ‘start-here’ example we will cover one of the most basic types of analysis performed after MD simulations: coordinate root-mean-squared deviation (RMSD, description of the command can be found here). It will also cover ‘tagging’ loaded topology and reference files in CPPTRAJ. RMSD measures the deviation of a target set of coordinates (i.e. a structure) to a reference set of coordinates, with RMSD=0.0 indicating a perfect overlap. RMSD is defined as:
![RMSD = \sqrt{ \frac{ \sum_{i=0}^N \left[ m_i \cdot (X_i - Y_i)^2 \right]}{M}}](https://s0.wp.com/latex.php?latex=RMSD+%3D+%5Csqrt%7B+%5Cfrac%7B+%5Csum_%7Bi%3D0%7D%5EN+%5Cleft%5B+m_i+%5Ccdot+%28X_i+-+Y_i%29%5E2+%5Cright%5D%7D%7BM%7D%7D&bg=ffffff&fg=000&s=0&c=20201002)
Where 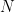
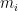
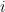
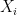
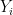
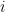
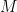
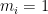

When calculating RMSD of a target to reference structure, there are two very important requirements:
- The number of atoms in the target must match the number of atoms in the reference.
- The ordering of atoms in the target must match the ordering of atoms in the reference.
For this example we will continue to use the same trpzip2 protein system. The trajectory is in NetCDF format, which is faster to process, more compact, higher precision, and more robust than the ASCII format. NetCDF is enabled by default in Amber, but if you find that your CPPTRAJ cannot read this trajectory please contact the Amber mailing list for help. The trajectory, associated topology, and other files can be downloaded here:
trpzip2.ff10.mbondi.parm7 – This is the topology file
trpziip2.gb.nc – This is the trajectory file
trpzip2.1LE1.1.rst7 – This is a restart file
trpzip2.1LE1.10.rst7 – This is a restart file
2GB1.pdb – This is a PDB file with a structure
Calculating RMSD
To start CPPTRAJ, type ‘cpptraj’ from the command line:
[user@computer ~]$ cpptraj CPPTRAJ: Trajectory Analysis. V6.4.5 (GitHub) ___ ___ ___ ___ | \/ | \/ | \/ | _|_/\_|_/\_|_/\_|_ | Date/time: 12/22/23 15:54:02 | Available memory: 936.329 MB
Load the topology (using the parm command) and trajectory file (using the trajin command):
> parm trpzip2.ff10.mbondi.parm7 [parm trpzip2.ff10.mbondi.parm7] Reading 'trpzip2.ff10.mbondi.parm7' as Amber Topology Radius Set: modified Bondi radii (mbondi) This Amber topology does not include atomic numbers. Assigning elements from atom masses/names. No SCEE section: setting Amber default (1.2) No SCNB section: setting Amber default (2.0) > trajin trpzip2.gb.nc [trajin trpzip2.gb.nc] Reading 'trpzip2.gb.nc' as Amber NetCDF Warning: NetCDF file time variable defined but empty. Disabling.
Specify the RMSD command:
> rms ToFirst :1-13&!@H= first out rmsd1.agr mass [rms ToFirst :1-13&!@H= first out rmsd1.agr mass] RMSD: (:1-13&!@H*), reference is first frame (:1-13&!@H*), mass-weighted. Best-fit RMSD will be calculated, coords will be rotated and translated.
In this case we are calculating mass-weighted RMSD using the rms command, saving to a data set named ‘ToFirst’, using all non-hydrogen atoms in residues 1 to 13, using the first frame as reference, and writing to a file named ‘rmsd1.agr’ (which will be written in xmgrace format due to the .agr extension). Note that by default the ‘rms’ command in CPPTRAJ calculates the best-fit RMSD, which means each structure is rotated and translated so as to minimize the RMSD to the reference structure (in this case the first frame). As such, the ‘rms’ command modifies coordinates for all subsequent commands unless ‘nofit’ is specified (more on that later).
Start the trajectory processing by typing run.
> run [run] ---------- RUN BEGIN ------------------------------------------------- PARAMETER FILES (1 total): 0: trpzip2.ff10.mbondi.parm7, 220 atoms, 13 res, box: None, 1 mol INPUT TRAJECTORIES (1 total): 0: 'trpzip2.gb.nc' is a NetCDF AMBER trajectory with coordinates, Parm trpzip2.ff10.mbondi.parm7 (reading 1201 of 1201) Coordinate processing will occur on 1201 frames. BEGIN TRAJECTORY PROCESSING: ..................................................... ACTION SETUP FOR PARM 'trpzip2.ff10.mbondi.parm7' (1 actions): 0: [rms ToFirst :1-13&!@H= first out rmsd1.agr mass] Target mask: [:1-13&!@H*](116) Reference topology: trpzip2.ff10.mbondi.parm7 Reference mask: [:1-13&!@H*](116) ----- trpzip2.gb.nc (1-1201, 1) ----- 0% 10% 20% 30% 40% 50% 60% 70% 80% 90% 100% Complete. Read 1201 frames and processed 1201 frames. TIME: Avg. throughput= 46462.1455 frames / second. ACTION OUTPUT: TIME: Analyses took 0.0000 seconds. DATASETS (1 total): ToFirst "ToFirst" (double, rms), size is 1201 (9.608 kB) Total data set memory usage is at least 9.608 kB DATAFILES (1 total): rmsd1.agr (Grace File): ToFirst RUN TIMING: TIME: Init : 0.0001 s ( 0.20%) TIME: Trajectory Process : 0.0258 s ( 87.61%) TIME: Action Post : 0.0000 s ( 0.00%) TIME: Analysis : 0.0000 s ( 0.00%) TIME: Data File Write : 0.0036 s ( 12.04%) TIME: Other : 0.0000 s ( 0.00%) TIME: Run Total 0.0295 s ---------- RUN END ---------------------------------------------------
Once the run has completed, if xmgrace is installed you can view the output right from the CPPTRAJ command line:
> xmgrace rmsd1.agr [xmgrace rmsd1.agr]

In this figure the X axis is the frame number and the Y axis is coordinate RMSD to the first frame (in Angstroms).
For more analysis examples, you can now follow the list of CPPTRAJ recipes available.